I. СИНТЕЗ КАНАБИДИОЛА
1. Введение
Из стеблей цветов женственного растения конопли (Cannabis sativa L.) может добываться зеленоватая смола, компоненты1) содержания которого вызывают сильное действие на центральную нервную систему. Это обозначается в зависимости от месторождения, как марихуана (Америка), гашиш (Средний восток), чараш (Индия) и т.д.
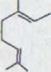
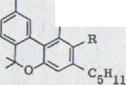
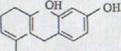
Сопоставление самых важных добываемых до сих пор из гашиша компонентов обозначено в схеме 1. При этом речь идет исключительно о фенольных связях, существующих в качестве Оливетола, точнее из Оливетолкарбонатовой кислоты и 10 атомов углерода, содержащих остаток углеводорода.
В данной работе будет описан синтез канабидиола2). Это вероятный биологический предшественник обеих физиологически активных связей ∆1,2-3,4-рыбьего жира-тетрагидроканнабинола и ∆1,6-3,4-рыбьего жира-тетрагидроканнабинола. Как показал уже более ранний опыт, (-)-каннабидиол через циклы катализирования кислотой может легко быть представлен в обеих последних двух связях3)4).
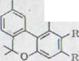
Схема1
R,=H R,=COOH
|
Каннабидиол I |
 Каннабидиол
– карбонатовая кислота II
Каннабидиол
– карбонатовая кислота II R=C5H11 R=C5H11(n)
|
Каннабидиол III |
 Каннабидиол – карбонатовая кислота IV
Каннабидиол – карбонатовая кислота IV |
∆1,2-3,4-тетрагидроканнабинол V |
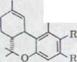 ∆1,2-3,4-тетрагидроканнабинолкарбонатовая
кислота VI
∆1,2-3,4-тетрагидроканнабинолкарбонатовая
кислота VI|
∆1,6-3,4-тетрагидроканнабинол VII |
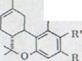
|
каннабигерол
VIII |
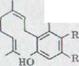 каннабигеролкарбонатовая
кислота IX
каннабигеролкарбонатовая
кислота IX|
каннабихром X
|
2. Теоретическая часть
2.1 Литературный обзор
Химическое исследование гашиша уходит корнями в середину прошлого столетия. В 1857 году указывали T. и Х. Смит5) в Эдинбурге, что физиологически активный принцип лежит в щелоченеразрешимом, высококипящем экстракте конопли и что речь идет не об алкалоиде. Wood, Spivey и Easterfield6) получали стекловидный продукт с постоянным варением, чисткой смолы, которую они называли каннабинол и рассматривали как унифицированную. Вплоть до появления исследовательских работ R. S. Cahn7) предполагали, что эта субстанция представляет активный принцип гашиша. Окончательная структура каннабинола доказывалась Адамсом8) и вскоре после этого Тоддом9) путем синтеза. Одновременно удавался Адамсу10) и сотрудникам отделение второй унифицированной, оптически активной субстанции из красного масла*), каннабидиол. Равная субстанция была изолирована Якобом и Тоддом11) из египетского гашиша. Она не обладает никакой физиологической активностью.
Первые тетрагидроканнабинолы производились в связи с разъяснением структуры и попытками синтеза каннабидиола. Таким образом, Адамс8) мог изолировать физиологически активные смеси от двойного изомерного соединения тетрагидроканнабинола, которые рассматривались позже Körte и Sieper12) через водоворотное распределение. Одновременно интересует также Тодда9) существование активных тетрагидроканнабинолей. Окончательный определение структуры материалов содержания гашиша, которое составляло долго большие трудности долго из-за плохих качеств кристаллизации и легкими изомерными двойными соединениями, удалось только после 1955 применением современных физических вспомогательных средств, как ядерный резонанс, массовая спектроскопия и газовая хромография. Как пример того - двойное соединение каннабидиола выявляется посредством НМР-Спектроскопией13), которое затем подтвердилось химическим опытом Körte14)15)
*) Дистилляцией экстракта конопли получают физиологически высокоспособствующую фракцию, так называемое " красное масло".
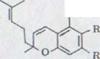
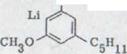
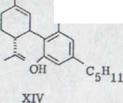
Первый синтез рацемического каннабидиола, даже хотя бы в очень незначительных объемах, описывался R.Mechoulam и Y.Gaoni16) в 1965 году (схема 2). Реакция цитрала с дериватом лития Олифетольдиметюлэтера XII, оборота с р-толуолсульфонилхлорида в пиридине и мерящая повторенная хроматография в алюминиевом оксиде и оксиде алюминия/нитрате бора ведет к около 3% выходу Каннабидиолдиметилл эфира XIII. Его можно синтезировать с избытком метилмагнияйодида в рацемическом каннабидиоле XIV.
Схема2
CHO OCH3
![]()
XI XII
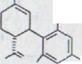 OCH3 OH
OCH3 OH
OCH3 C5H11
XIII
Körte15) описывал второй путь синтеза (схема 3). Конденсация алдола от масляного альдегида XVII с ацетоном ведет при одновременном отделении воды к замененному ядру Бензалакетона XVIII, которое может осуществляться в виде реакции XIX. Он реагирует в одной сопутствующей реакции с винил-метил-кетоном как неотъемлемый компонент к кетону XX. Новое масляное расщепление каннабиола-диметил эфира с метилмагниемсиумйодидоми и выделение такого полученного изомерного каннабидиола ведет к водородному распределению с 9,5% выходом каннабидиола.
Схема 3
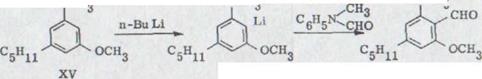 OCH3 OCH3 OCH3
OCH3 OCH3 OCH3
XVI XVII
(CH3)2CO
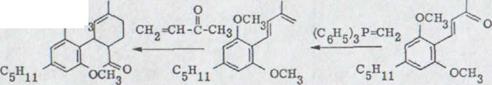 OCH3
OCH3
![]()
XX XIX XVIII
(C6H5)3P=CH2
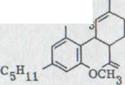
 OCH3
OCH3
XXI
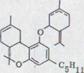
В 1964 выявилась17) первая унифицированная и одновременно физиологически активная связь из гашиша и к ней присоединилась при помощи NMR-спекции и патриархального синтеза структура V. Вторая активная связь, ∆1,6 изомерный VII получалась только из американской и мексиканской марихуаны. Синтез этой последней из двух связей в обобщенной форме описывался в том же году Тэйлором19) и Kierstead20). Особенно прост
Схема 4
![]()
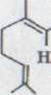
 С5H11 CHO
С5H11 CHO
![]() +
+
BF3-соединение


![]()
 XI
XI
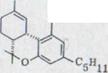
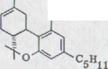
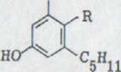
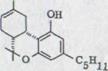 OH OH
OH OH
XXIV XXV XXVI XXVII
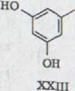
При этом синтез Тэйлора (схема 4). Обращение оливетола XXIII и цитрала XI с 10% BF3-составом ведет к смеси тетрагидроканнабинол изомера XXIV, XXV, XXVI, который можно раскалывать частями и иными препаратами гахроматографии. Получают кроме всего прочего (-)- ∆1,6- 3,4-переходного тетрагидроканнабинола с около 20% выхода. Однако, схему (-)-∆1,2-3,4-переходного тетрагидроканнабинола невозможно изобразить таким способом.
В дальнейших стали представляться также большим количеством аналоги тетрогидроканнабинола21-24). Может также показаться, что все признавали до сих пор двойное изомерное соединение тетрогидроканнабинола, а также соответствующую карбоновую древесную кислоту через ее массовую спектрацию, конечно, могли узнать25-27).
Физиологическая активность и биосинтез
Первые синтетические, физиологически активные тетрагидроканнабинолы исходят от рабочих групп, сплоченных вокруг Адамса и Тодда. В надежде найти связи между химическим строением и активностью гашиша, учеными рассматривалось большое количество родственных связей с тетрагидроканнабинолом. Тодд и последователи28) говорили уже в 1941 году, что, очевидно, положение гидроксилитных групп и разновидностей соединений алкилов зависит в большей степени от оливетольных частей. После более поздних исследований значимость двойного соединения и групп алкилов, кажется, будет существенна также в большей степени. Если цепь углеводорода варьируется, то оказывается максимум активности. Как наиболее эффективная связь описана позже Адамсом29) тетрагидроканнабинол со своими α, β-диметилсоединениями. Проверить активные субстанции пробовал Гайер тестом30) на кроликах и тестом на собаке31), причем, однако, очевидно, психотропное действие может испытываться только попытками только на человеке32).
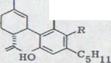
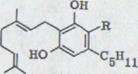
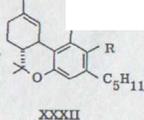
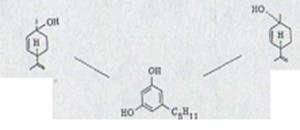
Проблема биогенеза гашиш - исходящих материалов широко поднималась уже у Тодда33). Сходство формул каннабидиола, тетрагидроканнабинола и каннабинола позволяли предполагать, что все эти продукты имеют общее происхождение из растения и одну конденсацию древесной кислоты (2,5,8-кратность) с олифетолом XXIII. Мечуламом и Гаони4) предполагалось биогенетическая последовательность, представленная на схеме 5. В качестве начального шага выступает предположительно образование каннабидиолкарбонатовой кислоты XXXlb, которая затем циклами переходит в тетрагидроканнабинол XXXIIb, который может переходить снова дегидрированием в инертный каннабинол.
Схема 5
 CH2OP2O63-
OH
CH2OP2O63-
OH
+
 |
XXVIII XXIX
 OH
OH
![]()
![]() XXX
XXXI
XXX
XXXI
 OH
OH
XXXIII
a) R=H
b) R=COOH
Крейси и Сантави35) и независимо от них Шульц и Хаффнер36) получили в 1958 году каннабидиоловую кислоту XXXIb, которая под декарбоксилиризацией легко может в переходить в каннабидиол. Последние из двух определили, что присутствует во всех исследованных ими сортах конопли каннабидиоловая кислота наряду с небольшим количеством "остаточного фенола", как главный материал, в то время как они нашли противоположные условия в гашише.
Эти результаты позволяли определить каннабидиоловую кислоту как материнскую субстанцию фенольных материалов. Керте и др1с). исследовали коноплю в течение вегетативного периода. Впервые они смогли подтвердить, что имеется в наличии в растении только каннабидиоловую кислоту. Каннабидиол и тетрагидроканнабинол – получают изолированно из конопли европейского возделывания - искусственные продукты. Активное вещество конопли южных широт необходимо приписывать следовательно исключительно климатическим факторам.
Другие работы
А. Примерный вариант синтеза.
Наибольшим в обзорах литературы обсужденным действием каннабидиола и тетрагидроканнабинола - это многоступенчатый синтез и ведение к изомерным смесям, из которых можно отделять только с трудом соответствующие рацемические продукты. Кроме того, эти действия незначительны. Происходящие в природе, оптически активные связи не могли до сих пор описываться. Здесь работает слишком описательный синтез, который основывается на следующих принципах:
1) Обе молекулярные части должны быть себе подобны и примыкающем классовым перемещением позволяют связываться.
2) При этом допускается в отличие от более раннего синтеза непосредственно выделение терпендеревата.
3) При правильном выборе исходного продукта вид двойного соединения может определяются заранее.
4) Применение оптически активного циклического терпена позволяет реакции связывать продукт и сырье.
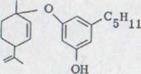
Молекула каннабидиола может быть разбита формально на 2 части, на полициклический терпен и оливетольный остаток (схема 6). Следовательно, требуется образование оливетолментадинил XXXIV для теплового режима и необходимости последовательного соединения.
Схема 6
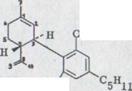
![]() OH
OH
 |
OH XXXIV
Она представляет перемещение легко доступных алилвинил системы, виниловая часть которой заменена оливетолом. Если цикличное перемещение реагирует на ее полукресельную форму, то получается для стерических последовательностей реакции следующая картина.
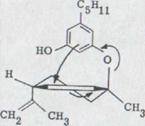 |
Fig. 1
Как известно, такие перемещения проходят строго стереоспецифически, как в случае перемещения Cope к Doering и при Claisen – перемещение, что доказывалось Хиллом, а также многочисленным синтезом в терпен- и стеройдустановках. Если оптически активный продукт конденсации структуры XXXIV может определяться, то как побочный продукт перемещения может явиться (+) или (-) -каннабидиол. Из связи XXXIV с 2 асимметричными атомами углерода теоретически возможны оба взаимных сочетаний, как представлено в схеме 7.
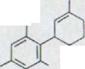 Схема
7
Схема
7
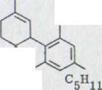 OH OH
OH OH
OH C5H11 HO
OH OH
XXXIV
Так как C-4 структурный атом не принимает участие в перемещении части терпена XXXIV, абсолютная конфигурация (-)-каннабидиола и вместе с тем тетрагидроканнабинола определяется тем, если централизованная связь в C-4, например, с (+)-глицеринальдегидом и если она известна.
Связи, которые исполняют долговые обязательства относительно доли терпена XXXI, были легко доступны в форме льда и переходного-p-метадинила-(2 - 8)-ole-(l); поступивших позже на их изготовление.
Для синтеза мягкий метод был необходим из за образования эфира. При этом необходимо было знание верификационных методов карбонатовой кислоты с многомерной ацеталией, как, например, N, N-диметилформамид – динеопентлацетал41)
Схема 8
![]()
![]() CH3 O---x
CH3 O---x
![]()
![]()
![]() RCOOH +
R`OH + N – CH
RCOOH +
R`OH + N – CH
CH3 O---x
RCOOR + 2HO---x + DMF
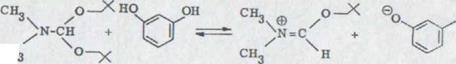
Эта реакция проходит при мягких условиях, не нуждается ни в каких кислых основных катализаторах и высоко используется в чистом сложном эфире. Находящиеся в незначительном избытке ацеталы делятся с водой на N, N-диметилформамид и соответствующий спирт. Если в схеме 8 заменяют указанные уравнения карбонную кислоту фенолом, то получают эфир фенола вместокарбонной субстанции. Следовательно, конденсация оливетола с соответствующими α, β спиртами привела бы к необходимому арилаллил эфиру XXXIV.
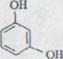
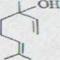
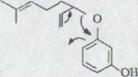
Так как такие эфиры были неизвестны, посредством реакции модели пробовали выполнить конденсацию фенолов из спиртов посредством N, N-диметил-формамид-динеопентилацетала. Как легко доступные исходные продукты выбирались резорцин и (±)-линалул.
Б.. Конденсация резорцина из леналула
Резорцин XXXV и леналул XXXVI освобождаются с 1,3-кратным количеством N, N-диметил-формамид-динеопентилацетала XXXVII в эфире под азотом при 48 градусах. После хроматографического расщепления при взаимодействии с водой продукты реакции силицагела после дистилляции изолировали в высоком вакууме и получили 12% масляный, тонкослойный хромографический унифицированный продукт. Так как резорцин является практически неразличимым в метилохлориде, здесь эфир должен был использоваться как решающее средство.
Связь указывает в IR. - спектре поглощение при 3600 см -1 (свободное OH выталкивающее колебание) и 3455 см -1 (связывающее ОH). Дальнейшие показатели - при 1622 см -1 для двойных соединений и, наконец, характерное сильное поглощение фенильного ядра при 1603 см -1 и 1510 см -1. Спектр NMR. показывает, что:
А) 2 ароматических протона эквивалентны δ =6,37ppm и в химических переносах отчетливо по-разному соотносятся к третьему при 6,95 ppm;
Б) Протоны метиловых групп в α - положении к ароматическим интенсивно определяются при δ = 3,3 ppm и только одним смежным водородом, что показывает ∆6 - двойное соединение;
2
В) ∆2 - двойное соединение существует.
Схема 9
 |
 |
||
![]()
![]() O-x
O-x
![]()
![]() +
+ N-CH
+
+ N-CH
![]() XXXV XXXVI XXXVII O-x
XXXV XXXVI XXXVII O-x
 |
||||||||
XXXVIII
Это состояние на схеме 9 указанных структур для 4-[3,7- диметилоктадия-(2,6)-yl] - резорцин XXXVIII в соответствии*). Спектр массы также соответствует указанной структуре (нромер молекулы: 246).
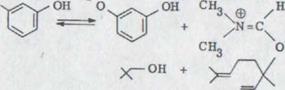
Ожидаемый сначала продукт конденсации, резорцинол-линалул эфир не получался. Возможность непосредственного прохождения реакции смеси при надстройке к выше описанному продукту невероятна в этом отношении, так как прохождения нуждаются в общем в более высокой температуре, что здесь, однако, никогда не нагревается даже до комнатной температуры. При реакции линалула с резорцином происходящие алкостические реакции устранения могут протекать как указано ниже:
*) О замещении резорцина см. также 24)
Схема 10

1) OH
CH3 x- OH
+
![]()
|
CH3
 HO
HO
3)
![]()
![]()
![]()
![]()
![]()
![]() CH3
CH3
XXXIX
При этом образовавшийся Иммониумион XXXIX может заменяться либо в SN1, либо SN2 реакции эфиром XXXIV. Вторая возможность - это непосредственная реакция резорцинаниона в SN1' или SN2' реакции с замененным ядром продукта XXXVIII, причем диметилформамид действует как хорошая обслуживающая группа. При применении оптически активных спиртов рацемического линалула реакция должна быть заключена просто в механизме и возможной стереоизбирательности.
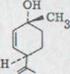
В. Конденсация резорцина с (+)-переходным – p – метадиеном – (2,8) – ol – (1)42)
Изготовление в дальнейшем использованной как исходные материалы цис- и транс-p-ментадиена-(2,8)-ole-(l)*) обсуждено немного. Экспонируется (+) лимоны XL под кислородной атмосферой в изопропил спирт и в присутствии стабилизируется, иак вещество O2 влияет на образовании героина из гидропероксидена. Они взаимодействуют с литийаллюминийгидридом или натрийсульфитом к соответствующим спиртам, из которых (+)-транс-p-ментадиен-(2,8 отделение)-ol-(l) XLI в 34% и соответствующий цис - изомер XL1I в 10% выхода фракционированной дистилляцией. Замечательным в этой реакции является, что вступление кислорода происходит исключительно в ∆1A - двойное соединение и при этом образованные продукты активны оптически. Получение оптической активности указывает, что реакция не управляет способными радикалами. Как механизм реакции принимают использование кислорода между соединением C H образования и одновременном перенесении AUyl поэтому. При этом все ожидаемые после этого принципа спирты образовываются применением аллил перемещением. :
Схема11
![]()
![]() 1) O2/Sens/hν
1) O2/Sens/hν
![]() + + + изомер
+ + + изомер
2)LIAL4
XL XLI XLII
В дальнейшем был найден более дешевый доступ к цис-спирту43)*). Он состоит в преобразовании (+)-∆4-каренов при комнатной температуре с концентрированной кислотой. При этом (+)-cis-ментадий -(2,8)-ol-(l) получается на выходе около 50%.
Оливетолу необходима вторичная исходная связь. Это изображалось на уже известном пути исследований Сатером и Вестоном45). Второй синтез представлялся Керте и Зипером12). Конденсация вышеупомянутых спиртов с резорцином и олевитолом описывается далее.
Преобразование резорцина с (+)-trans-p-ментадиеном-(2,8)-ol-(l) XLI вело к
*) Все использованные спирты стали поставляться фирмой Firmenich и Ко,
Laboratoire d'Etudes Proc£des, Ла-План, Женева.
*) Неопубликованные результаты Г. Охлофа между тем частично опубликовывают Голлник и Шаде44).
маслянистому продукту, который состоял в тонкослойном хроматографировании главным образом 3 компонентов. После хромотографии в силицагеле и дистилляции в высоком вакууме могут получиться следующие унифицированные связи (схема 12):
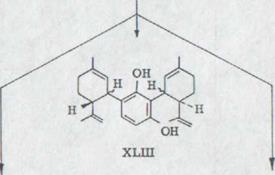
Как первичный продукт получили замененный 2 остатка ментадиенила резорцина XLIII с 6% выхода. Связь показывает в спектре IR. Колебания гидроксила при 3430 см -1, двойные соединения при 1640 и 1622 см -1 и о.о.p. - колебание деформации конечного постоянного винил протона при 890 см -1. В ядерном спектре резонанса получают АВ-систему для обоих ароматических протонов при 6,79/6,28 ppm. Оба олифинических C-2 водорода дают в итоге мультиэффект при 5,58 ppm, которые образуют конечный постоянный винил протон такой при от 4,9 до 4,3 ppm. Оба фениловых ядра α-постоянных протонов появляются как обширный неструктурированный всплеск при 4,2-3,2 ppm.
Связь унифицирована, правда, тонкослойным хроматографированием и активна оптически, однако, нельзя было решить, идет ли речь об изомерной смеси или нет.
Структура XLIV соответствующих связей могли определяться главным образом как масло с точкой кипения 150-160°, получающееся в 11% выхода. Она обнаруживает в спектре IR. характерные гидроксильные связи при 3500 (свободном ОH) и 3420 (связанный ОH) см -1. Двойные соединения поглощают при 1640 и 1618 см -1 конечные постоянные винил протоны при 895 см -1. Ядерный спектр резонанса показывает 2 разновидности ароматических протонов. C-5'- водород дает в итоге мультиэффект при от 7,12 до 6,83, C-4'-и C-6' водороды - такой при от 6,48 до 6,28ppm.
Схема 12
![]()
![]()
![]() HO
O -x
HO
O -x
![]()
![]() + + N-CH
+ + N-CH
O -x
XXXV
 OH OH
OH OH
OH
XLV
Олефинический протон появляется при 5, 5 ppm. Постоянный протон винила появляется при 4,68/4,58 ppm, который дает в итоге фенильное ядро α-постоянного протона при от 4,15 до 3,75 ppm. Так же массовый спектр (молекула 244) соответствует указанной структуре. Связь выявляет специфическое вращение от [α]D20 = -113° в спирте.
В 25% выходав соответствии с выше описанной связью получился 4-изомер XLV. Это может дистиллироваться также неразложимо в высоком вакууме при 180-190°/0,001. Спектр IR. (-)-4-[3,4-переходного p- ментадиена - 1,8-yl]- резорцин отличается от 2-замененной связи и др. характерным проявлением новой сильной связи при 1500 см -1. В спектре NMR. перенесение сигнала происходит для C 3 протона после воздействия более высокого поля: 3,6-3,15 ppm. (У 2-изомера от 4,15 до 3,75 ppm). Сигналы для ароматических протонов очень похожи своими связями XXXVIII.
Г. Конденсация оливетола с -trans- и (+)-cis-p-метадиеном-(2,8)-ol-(l)
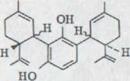
Смесь 6,2 mMol (+)-trans-p-ментадиена-(2,8)-ol-(l), 5,6 mMol оливетола и 7,3 mMol N, N – диметилформамид - динеопентилацетала в метилохлориде оставлялась 63 часа при комнатной температуре под азотом. После хроматографической обработки с водой получился продукт реакции, выделенные из силицагела и флоризила после дистилляции в высоком вакууме 3 тонкослойных хроматографированные унифицированные, маслянистые продукты (схему 13).
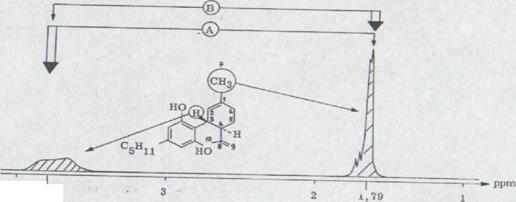
В 25% выхода получился (-)-каннабидиол I. Эта связь показывает при данном выходе*) характерную фиолетовую окраску. IR. Спектр показывает оба гидроксильных выталкивающих колебания при 3610 см -1 и 3450 см -1. Поглощение двойных соединений появляется как опора при 1640 см-1 и характеризует o.o.p. колебание деформации обоих конечных постоянных протонов винила при 890 см –1.
В NMR. Спектре**) выступают обамагнитных эквивалентных протона при 6, 2 ppm. Несколько распространенных связей при 5, 54 ppm соответствуют C 2 протону в двойном соединении. Оба винильных протона появляются как связи при 4,63 и 4, 54 ppm. Широкий мультиэффект при от 3,95 до 3,70 ppm может приписываться C 3 протону. Химическое перенесение после относительно низкого поля можно объяснять не только смежным финилином. Дополнительное действие происходит, очевидно, частично в олефиническом C 2 центре. Кроме того, нужно предполагать, что ароматическое кольцо, которое может вращаться свободно, лежит наиболее вероятно в уровне C 3 водорода, и последний из двух следовательно сильно проявлен46).
Оба венильных групп метилов появляются при 1,79 и 1,67 ppm; ώ-метиловой группы показывает характерную структуру толчка при 0,88 ppm.
*) Обрызгивают с 5% алкогольного KOH-решения.
**) См. выше также 13).
Схема 13
XXIII
![]()
![]() O -x
O -x
![]()
![]() N-CH
N-CH


![]() O –x
O –x
 |
OH
HO
I XLVII XLVI
В 100 Мгц ядерного спектрального резонанса фрагментация обеих связей могла наблюдаться при растяжении последовательности для обоих олефинических метилловых групп с мультиэффектом. Так как высокостепенное сцепление может ожидаться между C - 3 - водородом и C - 7 протоном метила, оно должно было бы выделяться посредством двойного эксперимента резонанса между обоими метилловыми группами.
Облучением частоты C - 3 протонов при 3,85 ppm получали вместо неструктурированного мультиэффекта при 1,79 ppm отчетливую, несколько широкую связь. Таким образом через C - 7 метил протон сигнал проходит при l, 79 ppm. Если облучение при l,79 ppm происходит наоборот, то его получают; согласно как и следовало ожидать влиянию мультиэффекта C-3 водорода, в то время как широкий двойной эффект будет очевиден (Рис. 2).
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() B
B
![]() A
A
Einstrahlung
Рис. 2
Сцепление обоих конечных постоянных виниловых водных материалов с C-10-метил протоном может разъединяться облучением при 4,7 ppm. Таким образом получают острый суммарный материал для C - 10 метиловой группы (Рис. 3).
![]()
![]()
![]()
![]()
![]() 1 Hz B
1 Hz B
 |
![]()
![]()
![]() A
A
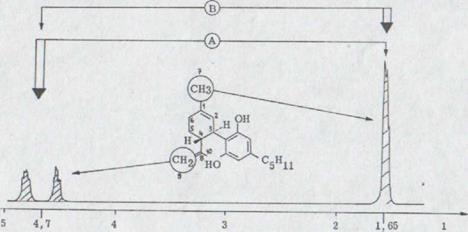
Рис. 3
Облучение частоты при 1,65 ppm (C - 10 метил) вызывает наоборот проспуск сигнала для конечного постоянного винил протона. Получают для нее АВ-систему с константой сцепления от 1 Гц.
Массовый спектр показывает молекулярные пики при 314. Характерную фрагментарность можно объяснять легко Retro-Diels-Alder- реакцией27).
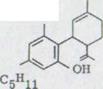
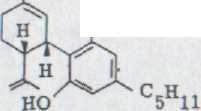
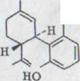
Как вторая унифицированная маслянистая связь получается (-)- cis- каннабидиол XLVI в 29% выхода. Связь отличается тонкослойной хроматографированностью сильно от многих полярных (-)- каннабидиолов и поэтому может хорошо отделяться хроматографически.
В данном случае связь показывает окраску цвета бордо. IR. Спектр является очень похожим на (-)- каннабидиола за исключением связей для протонов винила, которые появляются здесь при 895 см -1 и 887 см -1.
В спектре NMR., прежде всего, сигнал интересен при 3,75-3,35 ppm. Это отчетливо выявляется по отношению к ним при выходе (-)-каннабидиола после более высокого поля. Существенное различие лежит в дальнейшем в спектре UV., в то время как связь при 232 nm отложена в (-)-каннабидиоле после более короткой длины волн (221 nm). В массовом спектре оказывается более сильная интенсивность массы 246.
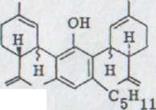
Наконец, в 5% выхода получали субстанционарный оливитол, к которому могла присоединяться на основании аналитических и спектроскопических данных структура XLVII. (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]- оливетол является конденсацией (-)- каннабидиола или по возможности cis-каннабидиола с двумя молекулами p- ментадинола. Несмотря на то, что субстанция тонкослойной хроматографированностью унифицирована, речь может идти о смеси стереоизомерных связей. Продукт - это масло, которое может дистиллироваться в высоком вакууме при 200°. Образуется коричневато-красная окраску в данном случае. В спектре IR. появляется гидроксил вытягивающее колебание при 3380 см -1, изолированные двойные соединения поглощают при 1640 см -1, и вытягивающее колебание для водорода выступают там ∆7- двойными соединениями при 895 см -1.
Водород в Ароматиках является в NMR. спектре сигналом при 6,23 ppm. Оба олефинических C - 2 протона дают возможность для мультисвязи при от 5,45 до 5, 7 ppm. Также как и в мультисвязи встречаются оба конечных постоянных винил протона при от 4,4 до 4,7 ppm. Оба C - 3 водорода обнаружимы неструктурировано между где-то 3,0-4,0 ppm.
(+)-cis-p-метадиен-(2,8)-ol-(l) в исходящих попытках за время реакции отдавал наряду с 64% олеветолом 21% (-)-каннабидиол и 5% стереоизомера (XLVI). Изменение растворителя (ацетонитрил вместо метиленхлорида) не дало в итоге никаких существенных различий в действии. Так же никакое повышение выхода продукта не могло получаться повышением температуры на 70° в бомбо-штурмовой трубе.
Выше изолированный (-)-каннабидиол был заменен с 3,5- динитробензуолхлоридом в пиридии соответствующим до 3,5-динитробензоатом10). Он мог получаться после чистки в силицагеле кристаллизовавшись и соответствовал точке эмали, смешанной пробе, специфическому вращению, IR. - и NMR. спектры с подлинной пробой до 3,5 динитробензоата13)*) от естественного (-)-каннабидиола в точном непосредственном сравнении. Расщеплением этого до 3, 5-динитробензоата с жидким аммиаком в толуоле оставался синтетический (-)-каннабидиол в кристаллизовавшей форме. Специфическое вращение, тонкослойное хроматографированное поведение, UV.-, IR.- и NMR. спектры оказывались идентичными с соответствующими данными пробы естественного каннабидиола**).
Соответствующий 3,5 динитробензоату (-)-cis-каннабидиол мог быть не кристаллизовавшимся.
Факт, что при всех этих реакциях первоначально ожидаемый эфир не мог ни изолироваться, ни устанавливаемое спектроскопически, пожалуй, не позволяет исключать его образование. Как исходит из вышеуказанных попыток, несколько больше каннабидиола всегда образуется при реакциях с резорцина или оливетола с trans-p-ментадиеном-(2, 8)-ol-(l). При единственно выполненной до сих пор попытке со спиртом противоположные условия выявились напротив в распределении продукта, а также установлена более медленная скорость реакции.
Механизм реакции, который был бы абсолютно соответствующим этим наблюдениям, является аномальным бималекулярным замещением. Эта SN2` реакция встречается обычно тогда, когда бималекулярное замещение затруднено α положением алилдеривата стеристическим препятствием, что здесь через третичный характер C1 углерода (в XLVTIII с * указано) в иммониумионе XLVIII. При этом
*) Профессор Р.Мехоулам, Hebrew University, Иерусалим, для предосталения сравниваемого препарата.
**) Профессор Ф. Кёрте, университет Бонн, благодарит за предоставление сравниваемого препарата.
 нуклеофил
входит на той же стороне молекулы, с которой высвобождается выходящая группа.
нуклеофил
входит на той же стороне молекулы, с которой высвобождается выходящая группа.
Рис. 4
Замещение транс и цис-спиртов в форме иммониумиона оливетолом воссоздано в схеме 14.
Схема 14
![]() OH
OH
H
XLVIII
![]()
![]()
![]() + H
+ H
![]()
 N= C – O
N= C – O
CH
IL
Так как входящая и выходящая группа должны быть копланарны и параллельны параллельны, возникает как видно из формулы XLVIII , можно получать из транс - спиртов мажоритарно cis - каннабидиол. В cis - спирте выходящая группа получает центральное положение. Он должен сначала из более нестабльной формы превратиться в форму с диаксиальной позицией имино - и изо пропилидовых групп. Равновесие будет лежать, конечно, на стороне стерически с значительно небольшой диэкваториальной соответственности; т.е. скорость замещения здесь будет медленнее, что в самом деле наблюдается. При этом образовывается больше (-)- каннабидиола, что происходит на схеме 14.
В ходе дальнейших исследований оказалось, что цис- или транс-p-ментадиен-(2,8)-ol-(l) непосредственно с оливетолом при использовании кислого катализатора (например, p – толуолсульфановая кислота) может заменяться Δ1,6)-3,4- транс-тетрагидроканнабинолом. Этот факт и в обоих случаях около 50% составляющий выход позволяют, однако, заманить в эту ловушку SN1'-механизм реакции, причем должен был образоваться образованный карбониумионом

![]() CH3
CH3
Рис. 5
по стерическим причинам (соприкосновение на противоположной стороне с изопропилидовой группой) продукт. Однако, окончательный вывод о ходе реакции будет возможен только посредством дальнейших опытов.
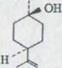
Д. Абсолютная конфигурация (-)-каннабидиола и тетрагидроканнабинола
Соединение конфигураций 2 здесь использованных алконолов было известно уже в работах Г. Охлофа и его последователей42). Таким образом катализированное гидрирование (+)-транс-р-метадиена-(2, 8)-ol-(l) XLI ведет к образованию транс-p-ментанолу-(l) L, конфигурация которого описывалась Хенбестом и МакЭльхинеем47) передачей β - Терпинеола из LI в L.
Навес и Грамполоф48) получали через выделение воды из транс – спирта и последующих сокращений образованного ментатриена LII с натрием спирт (+) -
*) Неопубликованные результаты Петрзилка и Сикемайера.
![]() лимонен
LIII.
Он принадлежит относительно к C-4-атому углерода ряда D- глицеринальдегидов49).
лимонен
LIII.
Он принадлежит относительно к C-4-атому углерода ряда D- глицеринальдегидов49).
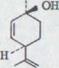
Схема 15
![]()
![]() 2H2 OH 1H2
2H2 OH 1H2
![]() XLI
L
LI
XLI
L
LI
AC2O/Δ
![]()
![]()
![]()
![]()
![]() Na/EtOH CHO
Na/EtOH CHO
![]() OH
OH
LII LIII CH2OH
Второй здесь использованный спирт мог образоваться стерическим соединением XLI только (+)-цис-p-ментадиена-(2,8)-ol-(l). Кроме того, его конфигурация доказывалась каталитическим гидрированием в цис-p-метоле-(l)48) LIV, который получается также обращением с 1 Mol H2 из соответствующего β - терпинеола LV. Связь с (+) -лимоненом проводилась аналогично, как при транс-спиртах.
Схема 16

![]() OH
OH
![]()
![]() 2H2 1H2
2H2 1H2
![]() XLII
XLII
AC2O/Δ
![]()
![]()
![]()
![]() CHO
CHO
![]()
![]()
![]() Na/EtOH
OH
Na/EtOH
OH
CH2OH
LII LIII
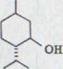
Так как теперь конденсация оливетола с (+)-cis-или (+) -trans-p-метадиеном-(2,8) -oL-(l) ведет к, конечно, кажущимуся (-)-каннабидиолу и C – 4 - центр во время реакции никакого изменения не претерпевает, абсолютная конфигурация (-)-каннабидиола и вместе с тем также тетрагидроканнабинола установлена согласно формулам I и XLVI. По номенклатуре Кана, Ингольда и Прелога50) I принадлежит к (3 R, 4 R) - конфигурации.
Все это было сделано уже в работе от Адамса и последователей51)(схему 17). Он указывал, что себе Тетрагидроканнабидиол LVI, который может получаться легко каталитическим гидрированием (-)-каннабидиола, с марганцовокислым калием в ацетоне окисляется в соответствующей метанкарбоновой кислоте LVII. Он превращался в соответствующий анилид. Он не доказал в итоге никакой депрессии пункта эмали, что сделал позже Курсанов52), из ментанола LVIII через метилхлорид LIX и последующей карбоксилизацией деривата произведенного сравниваемого препарата. Оптические вращения соответствующего анилида, однако не рассматривались. Здесь приведенные абсолютные конфигурации отличаются от более новых работ4) 13) 19) 20) 32) , где используются указанные на картинке формулы.
В появившейся недавно публикации Мешулама и Гаони53) при повторении работы Адамса получились те же результаты, что и у нас2).
Kat. OH
I
KMnO4/Ацетон
![]()
COOR
LVII
 1.)
Mg
1.)
Mg
2.) CO2
 |
![]() PCl5
PCl5
LVIII LIX
3. Экспериментальная часть
Общие замечания
Для определения нижеизложенных физических данных была выражена благодарность следующим ученым и их сотрудникам:
Господину В. Мансеру за анализы, которые получали в нашей микроаналитической лаборатории под его руководством.
Господину профессору доктору В.Симону за:
IR. Спектры: составлено с Перкин-Эльмер-Спектрографом (модель A 21, призма NaCl). Интенсивные подписки s,м,w сильно значимы, центральны, опорны. Соединение связей проводилось A. Д. Кроссом (54).
UV. Спектры: составлено с Beckmann-Spek-трохотрометром (модель десятичной системы классификации1). Заключенные в скобки ценности по максимуму поглощают ε-ценности.
NMR. Спектры: составлено с Varian-A 60 спектрометром (60 Мгц) тетраметилсиланом (δ= 0). Химическое перемещение составлено в ppm, взаимодействия Spin-Spin J в cps (Hz). Что значат s = Singlett, d = Dublett, t = Triplett, м = Multiplett и Sh = группу сигналов.
Господин доктор Й.Сайбль для тех
Массовые спектры, составленны с массовым спектрографом Hitachi RMU/6A, непосредственный доступ при 200° и 70eV.
Пункты эмали определялись в открытой трубочке в аппаратуре доктором Тоттоли и не исправлены.
Задачи точки кипения значат измеренные дистилляционными трубками температуры кипения,
Тонкослойная хроматограмма выполнялись " гелем кремния G тонкослойной хроматографии стали " (Merck), давление 2n/нормальное при около 120°/ ca.
Колониальная хроматограмма выполнялась с "гелем кремния" (Merck), 0,05-0,2 мм. Стоимость H2O указывается в хроматограмме; если никакой отметки нет, то гель кремния не дезактивировался.
 1.
4-[3,7-диметиллокладий-(2,6)-yl]- резорцин XXXVIII
1.
4-[3,7-диметиллокладий-(2,6)-yl]- резорцин XXXVIII
Смесь 1,1 g (10 mMol) резорцина, 1,7 g (11 mMol) только что дест. (t)-линалула и 3,0 g (13 mMol) N,N-диметиллформамид-динеопентилацитин в 20 мл эфира стала свободно реагировать под азотом и действием магнита при комнатной температуре. Сразу образовывался бесцветный осадок. Этот осадок образовывался в течение всего времени реакции. После 48 Std. смесь реакции принималась в 80 мл эфира, встряхивалась 3 раза с водой и 1 раз с раствором поваренной соли. Фаза эфира высушивалась сульфатом натрия и выпаривалась. Получалось 2,88g желтого масла). Оно далее хроматографировалось в 90g силицагеля. Графические тонкослойные хроматографические унифицированные фракции собирались и выпаривались.
Продукт mg
1. Бензол Не идентиф. смеси 272
Бензол 4-[3,7-диметилоктадий-(2,6)-yl]- резорцин XXXVIII 364
2. Бензол/
хлороформ 10:1 линалул 318
Бензол/
хлороформ 10:2 Не идентиф. смеси 631
3. Эфир резорцин 818
-----------
2403
![]() ______
______
Из фракции 1 c) 294 мг чистой субстанции могли добываться дистилляцией при 130°/0,001 Torr, что соответствует выходу 12%.
Sdp.: 130°/0,001 Torr
C16H22O2 Ber. C 78,01 H 9,00
Gef, C 77,97 H 8,90
UV. - Спектр (C2H5OH) λmax 281 (3,47)
IR. - Спектр (CHCl3): Связи при: 3600 (m), 3435 (m), 2920 (m), 1622 (s), 1603 (s), 1510 (s), 1453 (s), 1295 (m), 1147 (s), 1100 (m), 974 (s), 835 (m) см -1.
NMR. - спектр (CDCl3): δ =6,95 (м/lH),
6,37 (м/2H),
5,21 (м/2H),
3,30 (d/j=7Hz/2H),
2,14 (м/4H),
1,72 (d/J=3,5Hz/6H),
1,62 (s/3H) ppm.
Массовый спектр (2OO°/70, eV): молекулярный пик 246
2. Образование резорцина с trans-p-ментадиена-(2, 8)-ol-(l) XLI
Смесь 1,10 g (10 mMol) резорцина, 1,67 g (11 mMol) (+)-trans-p-ментадиена-(2,8)-ol-(l)*) и 3,01 g (13 mMol) N,N-диметилформамид-динеопентилацетала в 30 мл сухой смеси растворителя метиленхлорид/эфира 1:5 протекали при 72 Std. под азотом при комнатной температуре. При этом грубый в начале осадок оседал в итоге лишь частично. Смесь реакции обрабатывалась затем в эфире и встряхивалась 3 раза с водой (приложение небольшим раствором поваренной соли). Светло-желтая фаза эфира высушивалась сульфатом натрия и выпаривалась. Оставались 3,12 g светло-желтого масла. Эта недоимка хроматографировалась коллектором в 95 g силицагеле. Получили:
1. Бензол, 2 хлороформ и 3 эфир. Первая хроматография не дала в итоге никакого комплексного выделения:
[α]D18˚= + 73,6˚ (в субстанции)
Основной продукт: мг
1. a) неизвестный, согласно MS 3-замененный резорцин 65
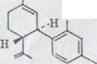
б) 2-замененный резорцин XLIII 425
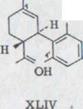
в) (-)-2-[3,4trans-p-ментадиен-(l,8)-yl]-резорцин XLIV 595
2. a) неизвестный 82
b) (-)-4-[3,4trans-p-ментадиен-(l, 8)yl]-резорцин XLV 884
3. резорцин 633
Характеристика продуктов реакции
a) (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-резорцин XLIII
 |
Фракция 1b хроматографировалась в 30-кратном количестве силицагеля с бензолом еще раз. После дистилляции в шаровой трубе при 210-220°/0,001 Torr получили 216 мг(5,7%) повисившийся унифицированный продукт.
Sdp.: 210-220°/0,001 Torr
[α]D20˚= -113˚ (c= 0,33/ C2H5OH)
C26H34O2 Ber. C 82,49 H 9,05
Gef, C 82,23 H 8,88
284 (3,3)
UV. –спектр (C2H5OH) λmax
232 (4,02) основа
IR. – спектр (CHCl3): Связи при: 3430 (s), 2920 (s), 1640 (m), 1622 (m), 1590 (m), 1485 (s), 1445 (s), 890 (s) см -1.
NMR. - спектр (CDCl3): δ =6,79/6,28 AB (J= 8,5Hz/2H),
5,58 (m/4H/2H через D2O взаимосвязи),
4,9-4.3 (m/4H),
4,2-3,2 (m/2H),
2,5-1.2 (m/22H), ppm.
Массовый спектр (2OO°/70, eV): молекулярный пик 378.
 б)
(-)-2-[3,4trans-p-ментадиен-(1, 8)-yl]-резорцин XLIV
б)
(-)-2-[3,4trans-p-ментадиен-(1, 8)-yl]-резорцин XLIV
OH
Фракция lc хроматографировалась еще раз в 30-кратном количестве силицагеля с бензолом. После дистилляции в шаровой трубке при 150-160°/0,001 Torr получили 258 мг(10%) унифицированного продукта.
Sdp.: 150-160°/0,001 Torr
[α]D20˚= -132˚ (c= 0,274/ C2H5OH)
C16H20O2 Ber. C 78,65 H 8,25*)
Gef. C 78,07 H 8,04
275 (3,1)
UV. – спектр (C2H5OH) λmax
281 (3,075)
IR. -спектр (CHCl3): Связи при: 3600 (s), 3420 (s), 2920 (s), 1640 (m), 1618 (s), 1585 (s), 1465 (s), 1280 (s), 1170 (s), 990 (s), 895 (s) см -1.
NMR. - спектр (CDCl3): δ =7,12-6,83 (m/1H), 6,48-6,28 (m/2H), 5,61 (s/1H), 4,68/4,58 (m/2H), 4,15-3,75 (m/расширенный/1H), 2,8-1,6 (m/11H), фон: 6,3-4,6 (2H), через D2O взаимосвязи), ppm.
Массовый спектр (2OO°/70, eV): молекулярный пик 244.
*)Вопреки повторенной дистилляции никакие лучшие выводы при анализа не получаться. Субстанция унифицирована тонкослойно хроматографически и ее спектроскопические данные соответствуют предположительной структуре.
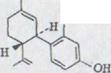
в) (-)-4-[3,4-trans-p-ментадиен-(l,8)-yl]-резорцин XLV
 OH
OH
Фракция 2c хроматографировалась еще раз в 30-кратном количестве силицагеля. После дистилляции в шаровой трубке при 180-190°/0,001 Torr получили 597 мг(24,5%) унифицированного продукта.
Sdp.: 180-190°/0,001 Torr
[α]D20˚= -113˚ (c= 0,225/ C2H5OH)
C16H20O2 Ber. C 78,65 H 8,25*)
Gef. C 77,75 H 8,08
281 (3,5)
UV. -спектр (C2H5OH) λmax
226 (4,0) опорный
IR. - спектр (CHCl3): Связи при: 3595 (s), 3430 (s), 2920 (s), 1640 (m), 1623 (s), 1595 (s), 1505 (s), 1460 (s), 1445 (s), 1165 (s), 975 (s), 895 (m) см -1.
NMR. - спектр (CDCl3): δ =6,95-6,75 (m/1H),
6,5-6,25 (m/2H),
5,65 (через D2O взаимосвязи)
5,12 (через D2O взаимосвязи)
5,50 (m/расширенный/1H),
4,68-4,59 (m/2H),
3,60-3,15 (m/1H),
2,50-1,40 (m/11H), ppm.
Массовый спектр (2OO°/70, eV): молекулярный пик 244.
В аналогично установленной попытке дистилировалась полученная при доработке недоимка (4,2 g) уже после испарения эфира в высоком вакууме. Дистиллят (2,5 g) хроматографировался как выше описано дважды в силицагеле. После дистилляции тонкослойно хроматографически унифицированной фракцией получили следующее распределение продукта:
Мг %
![]() дисубстантитуированный
резорцин XLIII 273 7.2
дисубстантитуированный
резорцин XLIII 273 7.2
(-)-2-[3,4-trans-p-ментадиен-(l,8)-yl]-резорцин XLIV 551 22.5
(-)-4-[3,4-trans-p-ментадиен-(l,8)-yl]-резорцин XLV 472 19.3
![]()
![]() резорцин
443 40.3
резорцин
443 40.3
![]()
![]() 1739 89.3
1739 89.3
3.Образование оливетола с trans-p-ментадиеном-(2,8)-ol-(l) XLI
1017 мг (5,64 mMol) оливетола и 944 мг (6,21 mMol) (+)-trans-p-ментадиена-(2,8)-ol-(l)**) опустили в 33 мл сухого метиленхлорида и 1700 мг (7,34 mMol) N, N-диметилформамид-динеопентилацин. Держали под азотом во время 63 Std./20° под магнитным воздействием.
Итог реакции получился затем в эфире и его встряхивали 5 раз с водой. Сначала эмульсия устранялась при добавлении удовлетворяемого целям хлорида натрия. След воды показал зеленоватую флуоресценцию.
Фаза эфира сушилась в сульфате натрия и выпаривалась. Получали 2,197 g желтого масла. Это дистилировалось главным образом при от 170 до 200°/0,001 Torr: 1,497 g. недовыход: 126 мг.
Дистиллят хроматографировался в 50 g силицагеля с бензолом. К концу хроматографирования получали 2% эфира. Нерасщепленные фракции собирались (504 мг) и во второй раз хроматографировались в 50g флоризила с гексан/5% бензолом. Получали следующее распределение в хроматографически очищенных дистиллированных субстанций:
Мг %
![]() (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
103 4
(-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
103 4
(-)-каннабидиол 440 25
(-)-cis-каннабидиол 514 29
*) См. сноску С. 42.
**)[α]D18˚= + 63,6˚ (в субстанции)
![]()
![]() оливетол
358 35
оливетол
358 35
![]()
![]() 1415 93
1415 93
Характеристика продуктов
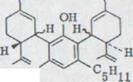 a) (-)-2,4-Bis-[3-p-ментадиен-(1,8)-yl]-оливетол XLVII
a) (-)-2,4-Bis-[3-p-ментадиен-(1,8)-yl]-оливетол XLVII
HO
Sdp.: 200°/0,005 Torr
[α]D18˚= -113˚ (c= 0,6/ C2H5OH)
лучевой тест: Коричневато-красная окраска
C31H44O2 Ber. C 82,98 H 9,89
Gef, C 82,90 H 9,88
211 (4,78)
UV. - спектр (C2H5OH) λmax
284 (3,45)
IR. - спектр (CHCl3): Связи при: 3420 (s), 2930 (s), 1640 (m), 1615 (s), 1575 (s), 1435 (s), 1375 (m), 1150 (s), 1090 (m), 1015 (m), 895 (s) см -1.
NMR. - спектр (CDCl3): δ =6,23 (s/1H),
5,93 (s/1H через D2O взаимосвязи),
5,75 (s/1H через D2O взаимосвязи),
5,59 (m/1H),
4,58 (m/2H),
4,0-3,0 (неструктурированный 1H),
2,8-0,9(m/33H) ppm.
Массовый спектр (2OO°/70, eV): молекулярный пик 448.
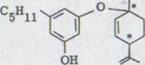
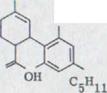
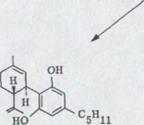
б) (-)-каннабидиол I*)
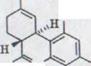 OH
OH
OH C5H11
Sdp.: 180°/0,005 Torr
Smp.: 66˚ (из гексана)***)
[α]D18˚= -129˚ (c= 0,45/ C2H5OH)
лучевой тест: Фиолетовая окраска
C21H30O2 Ber. C 80,21 H 9,62
Gef. C 80,27 H 9,71***)
С 80,25 H 9,62**)
UV. - спектр (C2H5OH) λmax 282/274 (3,07/3,90), опора 232 (4,15).
IR. - спектр (CCl4):
![]()
NMR. - спектр (CDCl3): δ =6,20 (s/2H), 5,54 (s, расширенный/1H), 4,63/4,54 (2s/2H), 3,95/3,70 (m/1H), фон: 6,4-4,0 (2H), через D2O взаимосвязи, 2,6-0,6 (m/22H) ppm.
Массовый спектр (2OO°/70, eV): m/e 314 (16%), 246 (34%), 231 (100%), 193 (10%),174 (8%), 121 (12%).
*) Спектроскопические данные кристаллизовавшихся проб.
**) Свободные, хроматографией и дистилляцией очищенные пробы.
***) Кристаллизовавшиеся, Bis-3,5-динитробензоатом очищенные пробы.
в)(-)-cis-каннабидиол XLVI
![]() OH
OH
HO
Sdp.: 180/0,005 Torr
[α]D18˚= -113˚ (c= 0,65/ C2H5OH)
лучевой тест: Окраска цвета бордо
C21H30O2 Ber. C 80,21 H 9,62
Gef. C 80,45 H 9,69
UV. -Spektrum (C2H5OH) λmax 282/274 (3,07/3,09), 221 (3,92).
![]() IR. -Spektrum (CCl4):
IR. -Spektrum (CCl4):
NMR. -Spektrum (CDCl3): 6,22 (s/2H), 6,06 (s/lH/через D2O взаимосвязи), 5,40 (s, расширенные/lH), 4,82 (s/lH/через D2O вхаимосвязи), 4,67 (s, расширенный/lH), 4,50 (s, расширенный/lH), 3,75-3,35 (m/lH), 2,9-0,7 (n./22H) ppm.
Массовый спектр (2OO°/70, eV): m/e 315 (13%), 314 (53%), 247 (18%), 246 (100%), 232 (18%), 231 (100%), 229 (10%), 207 (15%), 203 (33%), 193 (19%), 190 (15%), 189 (44%), 180 (11%), 176 (10%), 175 (66%), 174 (17%), 161 (25%), 160 (10%), 147 (21%), 137 (11%), 124 (10%), 119 (11%), 115 (11%), 107 (13%), 105 (11%).
4.Образование оливетола с cis-p-ментадиена-(2,8)-ol-(l) XLII
1000 мг (5,55 mMol) оливетола и 845 мг (5,55 mMol) (+)-cis-p-ментадиена-(2,8)-ol-(l) опускались в 40 мл сухого метиленхлорида и 1540 мг (6,66 mMol) N,N-диметилформамиддинеопентилацетал. Держалили под азотом во время 70 Std./20˚ под магнитным воздействием.
Итог реакции получился затем в эфире и его встряхивали 5 раз с водой.
Фаза эфира сушилась в сульфате натрия и выпаривалась при 20˚. Получали 2,197 g желтого масла.
Оно хроматографировалось в 70 g силицагеля с бензолом. К концу хроматографирования получали 2% эфира. Нерасщепленные фракции формулировались и еще раз хроматографировались. При этом получали следующее распределение хроматографически очищенных дистиллированных продуктов:
Мг %
![]() (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
145 6
(-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
145 6
(-)-каннабидиол 358 21
(-)-cis-каннабидиол 88 5
![]()
![]() оливетол
643 64
оливетол
643 64
![]()
![]() 1234 96
1234 96
5. Образование ацетонитрила
600 мг (3,33 mMol) оливетола и 0,556г (3,66 mMol) (+)-транс-p-ментадиена-(2,8)-ol-(l)*) опускались в 20 мл ацетонитрила и 1,0г (4,32 mMol) N,N-диметилформамиддинеопентилацетал. По истечении срока реакции 47 Std. при комнатной температуре получался эфир и его встряхивали 6 раз с водой. После сушения в сульфате натрия и выпаривании при 40˚ получали следующий выход тонкослойно хроматографических унифицированных продуктов:
Мг %
![]() (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
114 7,5
(-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
114 7,5
(-)-каннабидиол 230 22
(-)-cis-каннабидиол 310 29,5
оливетол 238 39,5
892 98,5
*) Согласно GC загрязнение из-за: 7% (+)-cis-p-ментадиен-(2,8)-ol-(l) и 3% (+)-trans-p-ментадиен-(2,8)-ol-(l) [α]D20˚= +73,6˚
6. Образование при высокой температуре
1,016г (5,64 mMol) оливетола и 0,946г (6,21 mMol) (+)-транс-p-ментадиена-(2,8)-ol-(l)*) и 1,70г (7,34 mMol) N,N-диметилформамиддинеопентилацетал расплавлялись с 25мл метиленхлорида в бомбо-штурмовой трубке и нагревались в масле во время 38 Std. до 70°. По истечении срока реакции получался метиленхлорид и его встряхивали 5 раз с водой. После сушения в сульфате натрия и выпаривания оставалось 2,162г, после дистиллирования в высоком вакууме при 190-200°/0,001 Torr: 1,154 g**). Далее это хроматографировалось в 30g силицагеля с бензолом. Получали следующие содержания в чистом дистиллировавшемся продукте:
Мг %
![]() (-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
89 4
(-)-2,4-Bis-[3-p-ментадиен-(l,8)-yl]-оливетол
XLVII
89 4
(-)-каннабидиол 374 23
(-)-cis-каннабидиол 401 25
оливетол 98 9,5
962 61,5
Bis-3,5динитробензоат от (-)-каннабидиол I
Смесь 730мг.(2,32 mMol) маслянистого (-)-каннабидиола и 1g (4,33 mMol) 3,5-динитробензолхлорида в 8 мл пиридина под 14 Std. взаимодействовали при исключении влажности при комнатной температуре. Затем нагревали результат реакции следующие 4 Std. на 60˚. На 10˚ охлажденный результат поливался смесью 15мл 1N соляной кислоты и 15 g льда, причем светло-желтый осадок не образовывался. Результат реакции оставляли в эфире и встряхивали 1 раз с 1N соляной кислотой и 2 раза с водой. Высушенная в сульфате натрия фаза эфира давала в итоге продукта 1,48g. После фильтрации в бензоле 6g силицагелем оставалось 1,38g маслянистого исходного материала. Он декристаллизировался из
*) Согласно GC загрязнение из-за: 7% (+)-cis-p-ментадиен-(2,8)-ol-(l) и 3% (+)-trans-p-ментадиен-(1,8)-ol-(l) [α]D20˚= +73,6˚
**) часть продукта превратилось в смолу, что не наблюдалось при образованиях при комнатной температуре.
метилацетата/метанола. Это получилось после двукратного декристаллизирования 610 мг (-)-каннабидиола-bis-3,5-динитробензоата в пункте 105-108˚. Из материнской щелочи в дальнейшем получилось 424 мг продукта (Smp. 101-102 °).
Sdp.: 105-108°*)
[α]D18˚= -77,5˚ (c= 0,4/ CHCl3)*)
C35H34N4O12 Ber. C 59,82 H 4,88 N 7,97
Gef. C 59,73 H 4,97 N 7,70
UV. – спектр (CHCl) λmax 251 (4,24)
IR. -спектр (CHCl3): Связи при: 3100 (w), 2920 (m), 1750 (s), 1635 (m), 1545 (s), 1455 (w), 1340 (s), 1250 (s), 1140 (s), 1070 (m), 990 (w), 925 (m) cm -1
NMR. - спектр (CDCl3)**): δ =9,32 (s/3H), 7,01 (s/2H), 5,34 (s/IH), 4,74/4,58 (s/2H), 3,59 (расширенный/d(J=ll герц)/1H), 1,77/1,58 (s,3H), 0,92 (s/3H) ppm.
Расщепление Bis-3,5-динитробензоата (-)-каннабидиолом I
547 мг (0,78mMol) Bis-3,5-динитробензоата отщиплялись от (-)-каннабидиола (Smp.104-107˚) в 2 мл толуола и проходили через 2 мл жидкого аммиака. Уже первые капли жидкого аммиака влекли за собой интенсивную красно-фиолетовую окраску смеси. Исход реакции был расплавлен при 41/2 Std. в бомбо-штурмовой трубке и оставлен при комнатной температуре. Затем аммиак испарился. Из коричневого остатка получалось после четырехкратного взаимодействия с петрол-эфиром (40-70°) 253 мг светло-желтого продукта. После фильтрации в бензоле 3 g силицагеля оставались 243 мг. Из этого смогли добыть двукратным декристализированием гексана 132 мг (54% теории) (-)-каннабидиола в гексагональных призмах призмах добываются (Smp. 66˚, оборот:-129°).
Проба дистиллировалась и давала в итоге снова оборот -129˚
II. СИНТЕЗ ЗАМЕНЕННОГО БУТЕНОЛИДА
1. Вступление и литературный обзор
Работы с замененным бутенолидом планировались в соответствии с планами доктора Петрцилка и выработанной попыткой синтеза цефалоспорина C I55). Изоляция этого продукта изменения материала цефолоспориума акремониума описывалась в 1956 году Абрахамом и Нютоном56a,b). С помощью химических57) и рентгенографических58) методов выяснилась в 1961 году структура этой связи. Немного позже открыли 6-аминопиницилиновую кислоту IV (схема 1), соответствующую 7-аминоцефалоспорановой кислоте II путем мягкого кислого гидролиза цефалоспорином C59). Замещением в аминогруппе также, как и в ацетоксиресте несущих метиленовых групп было возможно представить по связям весь класс цефалоспорина60,61), который сегодня используется в качестве антибиотиков в широком спектре действий.
Схема1
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() RCONH
RCONH
![]()
![]()
![]()
![]()
![]()
![]()
![]() O
O
III IV
|
![]() H3N
H3N
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
![]()
![]()
![]()
![]() OOC
OOC
O
COOH
I
Цефолоспорин C как характеристика β-лактам-дигидротиазиновой системы колец вместо β-лактам-тиазолидиновой системы имеет в своем составе пенициллин III. Путем синтеза прежде всего исследуют конструкцию этой необычной системы колец.
Первые попытки тотального синтеза62)63)64) берут начало от все того же образца синтеза пенициллина Шеханом65), в то время как уже пытались построить 3,6-дигидротиазиновую структуру, после чего позже долен был получиться ß-лактамовое кольцо. Таким образом Лове63) и одновременно также Турнер62) смогли впервые синтетически представить 3,6-дигидро-2H-l-тиазиновое кольцо (схема 2). Сложение Тиоацетамида VI с винил-кето сложным эфиром V дает в итоге дигидротиазин VII, который может при реакции с алюминиевой амальгамой переходить в тетрагидротиазин VIII. Дегидрирование в обезвоженной кислоте и высвобождение амина с бикарбонатом под одновременным изомеризацией двойного соединения ведет к желаемому 3,6-дигидро-2H-l,3-тиазина X.
Схема 2
![]()
![]()
![]()
![]()
![]()
![]() O CH2 S S
O CH2 S S
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() + CH3-C N
+ CH3-C N
COOCH2 Ph NH2 HO COOCH2Ph
V VI VII
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() S
S
S
S
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() HN H
HN
HN H
HN
COOCH2Ph HO COOCH2Ph
X VIII
Строительство β-системы лактамовых колец из 3,6-дигидротиазина в этом отношении затруднено, когда гетероциклическое кольцо содержит энамин, с гораздо более слабым нуклеофильным характером чем, например, вторичный амин кислоты пенициллина. Таким образом β-лактамовое кольцо цефалоспарина C пожалуй легко раскалывается, но непосредственный продукт гидролиза неизвестен. Его строение не дает возможность ему перенести даже мягкие условия его образования из лактама.
Интересный патриархальный синтез протекал через лилли-группу66) в то время как пенициллинсульфоксид мог превращаться в дигидро-тиазин-β-лактам.
Наконец, тотальный синтез удался Р. Б. Вудварду67)68) в 1965 году.
2. Теоретическая часть
Опыты попытки синтеза цефалоспорина C столкнулись со следующими частными проблемами (схема 3):
А) Изображение Δα,β-бутенолид-капьоновой кислота типа XI.
Б) Изображение cis- и trans-β-меркаплоакрелестера и его конденсация с органическими бромимдами; к схеме XII*).
В) Преобразование карбоксил- и изоцианатгрупп в бутенолид типа XI.
Схема 3
![]()
![]()
![]()
![]() HOOC
CH2Br
HOOC
CH2Br
![]() HOOC
HOOC
HOOC
HOOC
![]()
![]() CO O
CO O
CO O
CO O
XII XI
2.1 Изображение α-карбокси-Δα,β-бутенолида типа XI
Получению α-карбокси-β-метил-Δα,β-бутенолида XVI предшествовала известная Iизопропилидэнмалоновая кислота-диэтил сложный эфир (XIIIa)70) (Схема 4). Бромирование этого сложного эфира с 1 Mol-Aeq. бромсуццинимидом в тетрахлоруглероде при рассмотрении давало в итоге желаемую бромизопропилидэнмалоновую кислоту-диэтил сложный эфир XIVa, который мог к XVa лактонизироваться нагреванием на 150° при отделении А-этилббромида. Связь XVa - это дестилировавшаяся жидкость, который обозначена группами в спектре IR. при 1775 см -1(Δα,β –лактон с пятью кругами) и при 1705 см -1 (сложный эфир), а также
![]() CH3
CH3
![]()
![]() сигналами в NMR. Спектре
при δ=2,43 (s = ) и 4,78 (s/O
CH2) ppm.
сигналами в NMR. Спектре
при δ=2,43 (s = ) и 4,78 (s/O
CH2) ppm.
*) Карбоксильная функция этой карбонной кислоты должна позволять превращаться в изоцианатную группу. О синтезе β-лактамена из изоцианатов см. 69)