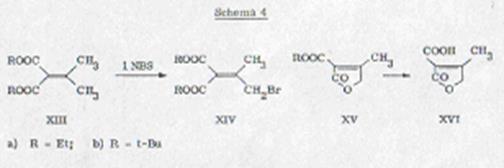
Гидролиз сложного эфира XVa с сконцентрированной соляной кислотой
привел к искомомой Лактонкарбоновой кислоте XVI, которая легко кристаллизовалась. Ее
структура является определяется числами анализа такими как спектр IR. (1785 см -1,
γ-лактон, 1705 см -1,
карбоксильная группа) и
спектр
 CH3
CH2
CH3
CH2
NMR. (δ= 2, 59 ppm (s/=/ )
и 4,92 ppm (s/= )
 Попытки непосредственного
субституирования в метиловой группе α-этоксикарбанол-ß-метил-Δα,β-бутенолида (XVa) не удавались. Реакция с 1 Mol Aeq. бромсуццинимида в тетрахлоруглероде
давала бромированный продукт, который в NMR-спектре дает сигнал при δ= 2,52 (s/=/CH3) И 7,29 (s/ C H) ppm., и структуру XVIIa определяется таким образом.
Попытки непосредственного
субституирования в метиловой группе α-этоксикарбанол-ß-метил-Δα,β-бутенолида (XVa) не удавались. Реакция с 1 Mol Aeq. бромсуццинимида в тетрахлоруглероде
давала бромированный продукт, который в NMR-спектре дает сигнал при δ= 2,52 (s/=/CH3) И 7,29 (s/ C H) ppm., и структуру XVIIa определяется таким образом.

 O Br
O Br
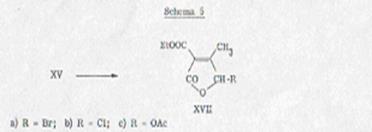
Изображение
аниона из XVa
обращением в хлорид натрия и преобразованием бромом давало в итоге продукт,
который совпадал всеми качествами выше описанного XVIIa, т.е. также здесь сибстутиент присоединился к кольцу.
Также хлорирование в сильно насыщенном протонами растворителе (серная кислота)
по Колоннитчу71) вела к внедрению хлора в кольцо лактона, что
сигналами в NMR. Спектре определяется при δ=2,5(s/=/CH3) и 6,54 (s/ C H)
ppm
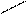
 O Br
O Br


 Наконец, также при обращении XVa со свинцом (IV)-ацетат остаток
ацетила входил в кольцов кольцо (δ=2,38(s/=/CH3) и 6,81 (s/ C H) ppm).
Наконец, также при обращении XVa со свинцом (IV)-ацетат остаток
ацетила входил в кольцов кольцо (δ=2,38(s/=/CH3) и 6,81 (s/ C H) ppm).
O OAc
Внедрение брома в метиловую группу удавалось не проводя обращение
изопропилидэнмалоновой кислоты-сложного эфира с 2 Mol-Aeq. N-бром-суццинимидом.
Эта реакция вела к симметричному дибромиду XIXb, который мог получаться
непосредственно из смеси реакции кристаллизировавшись (схема 6).
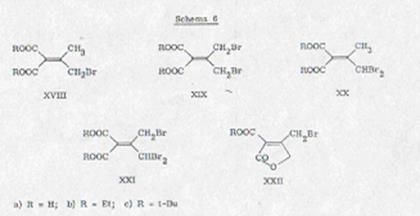
Лактонизация нагреванием, как она была проведена успешно
монобромидом XVIIIb, не
удавалась дибромидом XIXb; выходила при этом смола. Также при
обращении диэтил-сложным эфиром XIXb
с сконцентрированной серной кислотой при комнатной температуре в течение 20
часов, или ацетангидридом при обратной реакции, исходный материал стал
неизменно приобретать изначальную форму. Хлористый водород в диоксане вел к
возмещению брома хлором, без какой-либо лактонизации. Наконец,
она удалась при 48stdg.
нагревании сырой бромированной смеси с трифторной кислотой в бомбо-штурмовой
трубке и α-этоксикарбанил-ß-бромметил-Δα,β-бутенолид (XXIIb)
определялся в качестве масла; его структура определяется спектром IR. (1785 см -1
лактона, 1720 см –1 сложного эфира) и NMR. спектром (4,81 (s/-CH2Br) и 5,17 (s/-CH2-O-).
Как побочные
продукты выступили при этой реакции α-этоксикарбанил-ß-метил-γ-бром-Δα,β-бутенолид
(XIIa) и α-карбокси-ß-метил-Δα,β-бутенолид
(XIIa) (XVI).
XIIa мог бы возникнуть из наличествующего в сыром виде бромированной смеси
асимметричного дибромида
XXb; образование XVI следует приписывать также наличествующему монобромиду XVIIIb. Обе связи совпадали в их качествах с
подлинными препаратами (см. XIIa и XVI).
Так как при лактонизации трифторной кислотой полученный Aэтил сложный эфир XXIIb не мог быть без иного изменения
гидролизировавшись, чем с выше описанными последствиями реакции в виде
Di-t-бутил-сложного эфира.
Гидролиз
изопропилидэнмалоновой кислоты-диэтил сложного эфира и последующего продукта
сложного эфира с изобутиленом вел при правильных действиях к дибутил сложному
эфиру XXIb.
Бромирование с 1 Mol-Aeq. N-бромсуццинимидом в тетрахлоруглероде в
присутствии остатка бензолпероксида давало в итоге монобромид XVIIIc. Если проводилось бромироввание с 2 Mol-Aeq. N-бромсуццинимидом,
то получалась смесь бромированного продукта XVIIIc - XXc, как это было установлено
аналогично при диэтил сложном эфире. Искомый дибромид XIXc мог
чиститься хроматографией в силицагеле; все это находилось в бромированной смеси
в переменных количествах (40-70%).
При лактонизации смеси продуктов бромирования посредством
трифторной кислоты получался α-карбокси-ß-бромметил-Δα,β-бутенолид XXIIa,
который чистился хроматографией. Обнаружилось
выделение как t-бутиловой группы, так и лактонизация.
Если чистый дибромид XIXc был обработан трифторной кислотой, то наблюдалось только
расщепление t-бутиловой
группы при образовании дибромдикарбоновой кислоты XIXa, но никакой
лактонизации.
Тем не менее, в последствии оказывалось, что ощелочение и
лактонизация дибром-ди-t-бутил сложного эфира XIXc выполняется при еще мягких условиях. Если XIXc замещался в муравьиную кислоту с
серебротозилом, то последующее образование кристаллически давало
α-карбокси-ß-бромметил-бутенолид
(XI). При этом наверно группа бромида заменяется остатком тозилала и
расщипляется одновременно t-бутил
сложным эфиром. Структура является числами анализа, а также определяется
спектром IR. (1785 см -1 Δα,β-лактон и
1720 см -1, функция карбоксила) и NMR. спектром (δ= 4,81 s/-CH2Br
и 5,17 s/-CH2-O-) ppm.
2. Синтез cis- и trans-β-
меркапто-акрил сложного эфира и его конденсация с бромидом
Как саказано много выше72)-75), будут при
сложении тиолена в C=C группах,
очевидно, сначала образовывались cis-аддукты, которые переходят под влиянием кислоты, тепла,
излучения UV и т.д. легко в более стабильные trans-аддукты (к механизму s.76)). В противном случае
сверх этого ничто не было известно о сложении сероводорода с метил-сложным
эфиром пропиоловой кислоты. При первых попытках получали из метил-сложного
эфира пропиоловой кислоты с сульфидом натрия, гидроген сульфидом натрия или
солью сероводорода с tert.
аминами в метаноле соответственно диметилформамид после окисления
ди-(2-метоксикарбонил-винил)-сульфид, который возник, очевидно, при этом из 2
молекул первоначально образованного β-
меркапто-акрил сложного эфира, так как при преобразовании в смеси
реакции занесенной соли натрия с органическими бромидами (например,
бензилбромид) получали смесь дериватов cis- и trans-аддуктов
в различных условиях, в зависимости от условий реакции. При применении
натрийгидроген сульфида отношение cis-trans
составило 3:7, при преобразовании сульфидом натрия в присутствии лишнего натрия
- метилата напротив 6:4. Из смеси аддуктов легко можно было выявить транс-связь
вследствие ее относительно тяжелой растворимости. Чистая транс-кислота могла
получаться из транс-сложного эфира с щелочью питьевой соды.
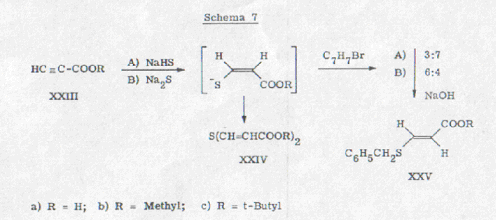
Cis-связь
можно было обогащать, правда, однако, таким образом мы ничего не выигрываем.
Однако, затем обнаружили, что чистый cis-аддукт
образовывается при сложении
бензилмеркаптана с пропиоловой кислотой.
Через сложно эфирное преобразование с дизометаном можно было
получить так же чистый cis-сложный
эфир (схема 8).
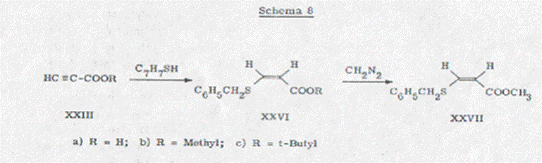
Из-за тяжелой растворимости солита натрия сероводорода в
почти всех органических растворителях пробовали проводить реакцию также с
солитом tert. амина. Если с H2S
получали удовлетворяемый выход пропиоловой кислоты-сложног эфира метила в эфире
или метаноле с каплей амина триэтила, то имела место быть экзотермическая
реакция, которая вела при лучшем исходе к вышеупомянутому сульфиду XXIV. Если
использовался, напротив, для этой реакции t-бутил сложный эфир пропиоловой кислоты, то состоялось данным лучшим способом нормальное
присоединение сероводорода в троекратное соединение, что могло показываться
снова преобразованием бензилбромида в бензил-спиртоксикарбонилвинил-сульфид
XXVI.
На основании всех этих опытов для реакции выбирали
α-карбокси-ß-бромметил-Δα,β-бутенолид XXIIa на следующих условиях:
Для выхода обезвоженного гидрогенсульфида натрия из сухого
диметилформамида связывали 1 Mol-Aeq. пропиоловой кислоты-t-сложног эфира бутила, до
-30˚ охлажденного выхода 1 Mol-Aeq. бромлактонкарбоновой кислоты XXIIa и, для нейтрализации действия
карбоксила, 1 Mol-Aeq, триэтиламина. Нормальное
окончание получения продукта реакции (см. экспер. часть) привело неожиданно к
хорошему выходу нейтрального тела, структура которого определяется, как XXIX
(схема 9). Первоначально образованная бутенолид карбоновая кислота XXIII была декарбоксилизированна по окончании опыта. Связь владеет в IR.- Спектре группой при 1575 см -1, которая, как обнаружилось,
для группировки является -S-CH=CH-CO- типической.

Чтобы различать формулы XXIX, XXIXa и XXIXb, субстанции модели
представлялись, как владеющие соответствующим хромофором. Преобразованием
аллилбромида (XXX) с ß-меркапто-акрил сложным эфиром-солитом натрия
(схема 10) получалась связь XXXI, которой соответствуют характерные для
конечной постоянной виниловой группы связи при 990 и 930 см-1 в IR.-спектре.
Через щелочезирование группы сложного эфира полученную
кислоту вела через действие калий-t-бутилата в диметилсульфоксиде к
изомеризированию XXXII78)79),
что спектре IR. Имеет типичную для trans- аэтилен связей группу при 950 см -1. Также NMR.-спектр связей XXXI и XXXII
является соответствующей предположительной структурой.
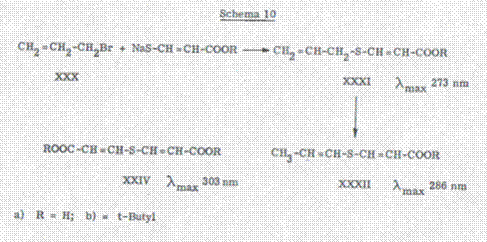
Связь XXXIa,
которая содержит систему
хромофоров XXIX и XXIXb,
владеет группой при 273 nm
в соответствие с UV.-пектром
XXIX (λmax 273 nm). Пропениловая связь XXXII,
которая владеет таким же хромофором, как и XXIXa, имеет максимум 286 nm . Вместе
с тем эта структура
может исключаться. Выше описанный ди-ß-акрил сложным
эфиром-сульфид XXIV
владеет хромофорной системой, которая напротив продлена XXXII и дальнейшими воздействиями
карбонила. В соответствии с этим максимум при 303 nm обнаруживается в XXIV. Между формулами XXIX и XXIXb различия найти смогли с помощь спектра NMR. и правил к
определению химический перенесения протонов в двойных соединениях после
Паскуал, Майер и Симон80).
Для винил протона связи XXIX
можно предвидеть
химическое перенесение около 6,0
ppm,
для него же в связи XXIXb
около 7,0 ppm.
Найденную группу при 6,1 ppm можно
таким образом хорошо отличать от структуры XXIX.
Дополнением при 0 ° при вышеописанной реакции
α-карбокси-ß-метил- Δα,β-бутенолида XXIIa с солью натрия
α-меркапто-сложного эфира акриловой кислоты, полученный продукт мог
значительно избегать декарбоксилизации, и таким образом получали искомую
карбоновую кислоту XXVIII в
аморфной форме, которая переходила, тем не менее, уже при комнатной температуре
медленно при убывании угледиоксида в XXIX
3. Преобразование карбоксильной и изоцианатной функций в бутонолиды
типа XI
Для запланированного преобразования дикарбоновой кислоты XII
в соответствующий изоцианнат стал легко доступно
использоватьα-карбокси-ß-метил-Δα,β-бутенолид
(XVI) как субстанцию
модели. Обращение фосфорпентахлорида в метиленхлорид привело к соответствующему
хлориду кислоты XXXIII (схема 11), которая давала эквивалентный натрийацид или
калийацид в ацетоне и соответственно ацетон/водe в хорошем выходе ацида кислоты XXXIV.
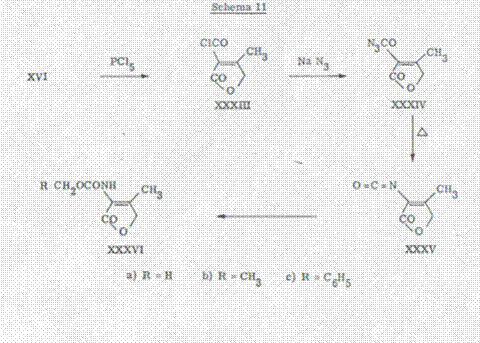
Если обыкновенный избыток(остаток) натриумацида
использовался напротив для выше описанной реакции, вместо нейтрального ацида
XXXIV получали таким образом кислоту, которая показывала широкую группу при
3000 см -1 и типичную для функции ацида группу при 2120 см –1 в
спектре IR., напротив никакая группа лактона не обладала при 1785 см -1.
Очевидно, лишний натриумацид реагировал не только с хлоридом кислот, но и
открыл кольцо лактонов.
Короткое нагревание ацида XXXIV в толуоле вело к изоцианату, который можно было
идентифицировать преобразованием в хорошо-кристаллизовавшийся уретан XXXVI. их числа анализа, IR. - и NMR. спектры хорошо
согласованы с предпологаемой структурой.
3. Экспериментальная часть
( Общие замечания см.
с. 38)
Монобромизопропилид-малоновая
кислота-диэил сложный эфир XIVa
При взаимодействии с лампой 200W в обратном течении варились 20g (0,1 Mol)
зопропилидмалоновая кислота-диэил сложный эфирa,
19 g
(0,11 Mol)
N-бросуццинимида
и 200 мг
бензолпераксида в
тетрахлоруглероде 2 Std.
Затем выпаривался сверху плавающий коричневатый выход натриумтиосульфата,
фильтрат с разбавленным выходом натриумтиосульфата всряхивался в вакууме: 28g. После
тонкослойной хроматограммы*) речь
шла о смеси из монобромида и небольшой исходной субстанции. В ходе анализа
дважды хроматографировалась в силицагеле с гексан/бензолом и затем при
100-110˚ дистиллировалась.
C10H15O4Br Ber. C 43,03 H 5,42
Br 28,63
Gef. C 43,08 H 5,49 Br 28,62
Спектр IR.
(CHCL3): группы и т.п. при: 2950
(m), в 1725 (s), 1640 (m), 1250 (s), 1150 (s), 1060 (s)см
-1.
Спектр NMR. (CDCl3): δ = 1,30 (t/J = 7 Hz/CH3-CH2-),
2,15 (s/=/CH3), 4,24 (q/J = 7 Hz/CH3-CH2-),
4,28 (s/=/CH2Br) ppm.
α-этоксикарбанол-ß-метил-Δα,β-бутенолид (XVa)
и α-карбокси-ß-метил-Δα,β-бутенолид XV
a) Бутенолид XVa
26
g
(0,093 Mol)
монобромизопропилида-малоновой кислоты-диэтил сложного эфира нагревались
1 Std. на 150-160°/12 Torr. При этом могло
наблюдаться образование Аэилбромида. Нехватка
(около 17 g) в дистиллировалась в HV.:
1. Фракция, 1,31g, 105-110°/0,001 Torr, исходный продукт; 2 фракция, 8,20g,
140-145°/0,001 Torr,
бутенолид через (52%); остаток, 2,74g, смолянной.
Спектр IR.
(CHCL3): 2950 (m), 1775 (s), 1705 (s), 1650 (m), 1325 (m), 1040 (s) cm-1.
*) бензол/10% эфира, доказательство посредством
0,2-proz. перманганата калия.
Спектр NMR.
(CDCl3): δ = 1,38 (t/J=7Hz/CH3-CH2-),
2,43 (s / =/CH3),
4,34 (q/J = 7Hz/CH3-CH2-), 4,78 (s/-CH2-O-) ppm.
б) Бутенолид XVI
Нагревали 300 мг (1,76 mMol) XVa с 2 мл
концентрированной соляной кислоты в 2 мл диоксана 45 минут до 85°. При
выпаривании в вакууме кристаллизовался непосредственно XVI. Из ацетона/петролэфира
кристаллы при Smp. 138-139°.
C6H6O4
Ber. C 50,71 H 4,26
Gef. C 50,79 H 4,30
Спектр IR. (CHCL3): группы и
т.п. при: 3100 (b), 1785 (s), 1705 (s), 1640 (m), 1275
(s), 1045 (s) см -1.
Спектр NMR.
(CDCl3): δ
= 2,59 (s/=/CH3), 4,92 (s/-CH2-O) ppm.
α-этоксикарбанол-ß-метил-γ-бром-Δα,β-бутенолид (XVIIa)
а) 1,70 g
(10 mMol) XVa и 1,86 g N-бромсуццинимида (ber. для 10 mMol 1,78 g) варились в 25 мл тетрахлоруглерода
при воздействии 200-W-лампы
1 Std. под обратным
течением. Затем суццинимимд фильтровался, фильтрат с выходом натриумтиосульфата встряхивался в
вакууме один раз и выпаривался: 2,07 g. Проба дистиллировалась при 110-130 /0,001 Torr.
б) 850 мг (5 mMol)
XVa выходило в 10 мл
диметоксиэтан (дестиллировалось через гидрид натрия и под азотом с 120 мг
гидрида натрия (250 мг 50-proz.
дисперсия гидрида натрия в масле перемешивалась с толуолом, фильтровались, и
использовался еще толуолом - овлажненный гидрид натрия). Этот выход опускали
постепенно в 5 мл 1M
выхода брома в хлороформе и держали 5 минут при 20°. Затем распределяли между
метиленхлоридом/водой одновременно несколько выходов натриумтиосульфата с целью
получения органического выхода. После высыхания органической фазы под сульфатом
натрия и выпаривания оставалось 1,03g, которые хроматографировались в
силицагеле с бензолом: 850 мг (68%), которые совпадали во всех качествах с под
a) описанным продуктом.
Спектр IR. (CHCL3): группы и т.п. при: 2960 (ra), 1785 (s), 1705 (s), 1640 (m), 1230 (s), 1015 (s), 805 (m) см -1.
Спектр NMR. (CDCl3): δ =1,39 (t/J=7Hz/CH3-CH2-), 2, 52 (s/=/CH3),
4,39 (q/J=7Hz/CH3-CH2-), 7,29 (s/0—-C-H-Br)
ppm. Обращение(лечение)
в льдяной уксус/бромовую кислоту во время 18 Std. при 20˚ не давало в
итоге никаких изменений в исходном продукте.
α-этоксикарбанол-ß-метил-γ-хлор-Δα,β-бутенолид
(XVIIb)
б) 850 мг (5 mMol)
α-карбэтокси-ß-метил-Δα,β-бутенолид
добавили к 15 мл 100-proz. Серной кислоты и обработали 5 Std. под воздействием магнита сухим
газообразным хлором с 1000-W-UV.-лампой. Затем вытряхивали с метиленхлоридом и
льдом. Полученные 673 мг субстанции хроматографировали силицагелем с бензолом.
Наряду с небольшим исходным продуктом получили 350 мг бутенолида VIIIb в качестве
масла.
Спектр IR.
(CHCL3): группы и т.п. при: 2960 (ra), 1785 (s), 1720 (s), 1660 (ra),
1245 (m), 1025 (m)см -1.
Спектр NMR. (CDCl3): δ =1,41 (t/J=7Hz/CH3-CH2-), 2, 50 (s/=/CH3),
4,39 (q/J=7Hz/CH3-CH2-), 6,54 (s/0—-C-H-Br)
ppm.
Связь
охарактеризована сигналом (s, 3H) при 2,5 и (s, lH) при 6,54 однозначно как
α-этоксикарбанол-ß-метил-γ-хлор-Δα,β-бутенолид.
α-этоксикарбанол-ß-метил-3-ацетокси-Δα,β-бутенолид
(XVIIc)
342 мг (2 mMol)
α-этоксикарбанил-ß-метил-Δα,β-бутенолид
смешивались с 950 мг свинцового тетрааццетата и 10 мл бензола, который
нагревали водяной баней и оставляли на ночь. Возникший коричневый осадок
фильтровался, с метилхлоридом/водой со льдом вытряхивался и при 120-130/0,001 Torr дистиллировался, причем
получилась смесь исходного материала с XVIIc.
Спектр IR.
(CHCL3): группы и т.п. при: 2940 (m), 1785 (s), 1720 (s), 1640 (m), 1245
(m), 1020 (m) см -1.
Спектр NMR. (CDCl3): δ =1,38 (t/J=7Hz/CH3-CH2-), 2,21 (s/CH3-CO),
2,38 (s/=/CH3), 4,38 (q/J=7Hz/CH2-CO), 6,81 (s/0—-C-H-OAc)
ppm.
Связь
охарактеризована сигналом δ
=2,38 и 6,81 однозначно как
α-этоксикарбанил-ß-метил-γ-ацетокси-Δα,β-бутенолид.
1,3-дибромизопропилид-малоновая кислота-диэтил сложный
эфир (XIXb)
20 г (0,1 Mol)
изопропилид-малоновая кислота-диэтил сложного эфира и 38,2 г N-бромсуццинимида (0,11 Mol) варились
тетрахлоруглероде при воздействии лампой 200W 7 Std. под обратным течением. Затем суццинимид фильтровался, фильтрат
с выходом натриумтиосульфата встряхивался, в вакууме выпаривался и высушивался
масляный остаток при 60°. В холодильнике продукт кристаллизовался. Из эфира
петрола 14,5 g (40%), Smp. 47- 48°.
C10H14O4Br2 Ber. C 33,54 H 3,94 Br 44,64
Gef. C 33,49 H
3,80 Br 44,49
Спектр IR.
(CHCL3): группы и т.п. при: 2905 (w), 1695 (s), 1610 (w), 1245 (w) см -1.
Спектр NMR. (CDCl3): δ = 1,32 (t/J = 7 Hz/CH3-CH2-),
4,3 (q/J = 7 Hz/CH3-CH2-O), 4,43 (s/-CH2Br)
ppm.
α-этоксикарбанол-ß-бромметил-Δα,β-бутенолид
(XXIIb)
2,0 g дибромизопропилид-малоновая кислота-диэтил сложного
эфира XIXb нагревались
с 8,0 мл трифторной кислоты 48 Std.
при 80˚ в бомбо-штурмовой трубке. Затем образованный аэтилбромид и
трифторная кислота дистиллировались в вакууме. Остаток (1,678g)
хроматографировалсяв 50g силицагеля с бензолом. Как основной продукт 800 мг
бутенолида XXIIb (53%)
получился как масло.
C8H9O4Br Ber. C 38,58 H 3,64 Br 32,09
Gef. C 38,78 H 3,54 Br
32,17
Спектр IR.
(CHCL3): группы и
т.п. при: 2980 (s), 1785 (s), 1720 (s), 1635 (s), 1355
(s), 1050 (s)см -1.
Спектр NMR. (CDCl3): δ = 1,43 (t/J = 7 Hz/CH3-CH2-),
4,48 (q/J = 7 Hz/CH3-CH2-), 4,81 (s/-CH2Br),
5,17 (s/-CH2-O-) ppm.
Изопропилид-малоновая
кислота-ди-t-сложный
эфир бутила XIIIb
42g (0,29 Mol) изопропилид-малоновой
кислоты и 160 мл диоксана смешали в
500 мл поршне при охлаждении льдом с 8 мл 100-proz. серной кислотой. Теперь при
от -10 до -15˚ изобутилен оставили при соприкосновении с магнитом до тех
пор, пока объемы возросли на около 1/3, и позволял оставили затем под твердым
герметичным стеклом на 12 Std.
при 20˚. Чтобы сместить равновесие по возможности на сторону сложного
эфира, второй раз он охлаждался и появился бутилен. После дальнейших 6 Std. смешивался с эфиром, встряхивался
в щелочи питьевой соды и в воде со льдом, сушился в сульфате натрия и
выпаривался вращением при 40˚: 49,96g масла. Для анализа проба
дистиллировалась при 140˚/0,001 Torr.
C14H24O4 Ber.
C 65,59 H 9,44
Gef. C 65,39 H 9,47
Спектр IR. (CHCL3): группы и т.п. при: 2990 (m), 1720 (s), 1645
(без
растворителя) (w), 1370 (m), 1250 (s) см -1.
Спектр NMR. (CDCl3): δ = 1,53 (s/C(CH3)3),
2,04 (s/=/CH3) ppm.
Из спиртовой субстанции после заквашивания образовалась
экстракцией соляной кислоты с эфиром 16,5g смесь изопропилид-малоновой кислоты и ее бутил –
полусложный эфир. Эта фракция стала соответственно без отделения снова
эфироваться, вследствие чего выход повысился на 80%.
Бромизопропилид-малоновая
кислота-ди-t-сложный
эфир бутила XIIIc
С 5g (19,5 mMol)
изопропилид-малоновой кислоты-ди-t-сложного эфира бутила и 100 мл тетрахлоруглерода
связали 3,6g (20,3mMol) N-бромсуццинимида и 250 g
бензоулпероксида. Варили смесь при экспозиции 1 1/2 Std. под обратным течением, причем выход
был снова желтоватым; в на дне раствора плавал суццинимид. Смесь охлаждалась,
суццинимид растворился, его с 400 мл тетрахлоруглерода разбавили, 3 раза с 100
мл ледяным выходом гидрогенкарбонатом натрия встрясывался, с водой со льдом
вымывался и в сульфате натрия высушивался. Растворитель удалялся при водяной
бане при 40˚ в вакууме струями воды и выход высушивался при HV.: 6,5g, которые согласно
тонкослойной хроматографии содержали (бензол/А-эфир здесь 10:1, очевидно
0,2-proz выхода перманганата калия) наряду с искомым монобромидом только
небольшой исходный продукт. Путем хроматографии из 4g исходного материала в
120g силицагеле с бензолом 1,593g (30,5%) получали чистый монобромид XVIIIc в качестве масла.
n20D=1,4760
C14H23O4Br Ber. C 50,16 H 6,92 Br 23,84
Gef. C 50,09 H 6,74 Br 23,73
Спектр IR. (CHCL3): группы и т.п. при: 2990 (m), 1730 (s), 1645 (m), 1395 (m), 1380 (s), 1280 (s), 1155 (s), 1070 (m) см -1.
Спектр NMR. (CDCl3): δ = 1,57 (s/-C(CH3)3),
2,10 (s/=/CH3), 4,28 (s/CH2Br) ppm.
1,3-дибромизопропилид-малоновая кислота-ди-t-бутил сложный эфир XIXc
24,1g (0,094 Mol)
ди-t-бутил сложный эфир XIIIb, 33,5g (0,188 Mol) N-бромсуццинимид, 500 мг
бензиолпероксида и 450 мл тетрахлоруглерода варились в 1-1-круглой колбе в
холодильнике и хлоркальциевой трубке при экспозиции с лампой 200W 1 1/2 Std., где суццинимид плавал сверху.
Холодный выход взаимодействовал с суццинимидом, с выходом гидрогенкарбоната
натрия и воды со льдом встряхивался, в сульфате натрия высушивался и при
40˚ вращением выпаривался: 39,03 g (99% чистый дибромоксида XIXc, как
расчитано). Этот дибромид использовался соответственно без дальнейшей очистки
для лактонизации. Проба хроматографировалась в силицагеле с бензолом и
составляла таким образом 40% выход чистого 1,3-дибромизопропилид-малоновая
кислота-сложный эфир бутила в качестве масла.
n20D=1,4983*)
C14H22O4Br2 Ber. C 40,60 H 5,35 Br 38,59
Gef. C 40,53 H 5,32 Br 39,08
Спектр IR.
(CHCL3): группы и т.п. при: 2940 (m), 1705 (s), 1620
(без
растворителя) (w), 1140 (s) см -1.
Спектр NMR. (CDCl3): δ = 1,56 (s/-C(CH3)3),
4,47 (s/CH2Br) ppm.
α-карбокси-ß-бромметил-Δα,β-бутенолид (XIIa) из 1,3-дибромизопропилид-малоновая кислота-ди-t-сложный эфир бутила XIXc
а) Посредством трифторовой кислоты
10g XIXc
нагревались с 30 мл трифторовой кислоты в бомбо-штурмовой трубке 48 Std. при 80°. После выпаривания
6,104g (ber. 5,34g)
получали коричневый продукт, хроматографировался в 130g силицагеля с бензолом. В качестве
основного продукта получили 2,107g маслянистого бромлактона XXIIa.
При обращении XIXc
с трифторовой кислотой при gew.
темпе дибромизопропилид-малоновая кислота XIXa: 1,35g (0,33 Mol) XIXc оставалась только 1,3 мл трифторовая кислота при комнатной
температуре. После 1/2 Std.
образовывались первые кристаллы, и после 3 Std. реакция была закончена. Трифторовая кислота вымывалась и
получались кристаллы 2 раза 1,3 мл тетрахлоруглерода. 1 фракция: 0,8g = 81%, Smp. 143 °. Из материнской щелочи
*) Как
побочные продукты хроматографировались при обращении следующие бромиды и
представлялись при этом:
a) 1,1,3-трибромизопропилид-малоновая кислота-t-сложный эфир
бутила XXIc, из эфира петрола выкристаллизировались, Smp. 107 °.
C14H21O4Br3 Ber.
C 34,10 H 4,29 Br 48,62
Gef. C 34,12 H 4,36 Br 48,20
Спектр IR. (CHCL3):
группы и т.п. при: 2960 (m), 1710 (s), 1620
(w), 1370 (s), 1285 (s), 1155 (s) см -1.
Спектр NMR. (CDCl3): δ = 1,58 (d/J=1,5 Hz/-C(CH3)3),
4,56 (s/=/CH2Br), 7,32 (s/=/CHBr2) ppm.
b) 1,1-дибромизопропилид-малоновая
кислота XX, выкристаллизировавшаяся из бензола, Smp. 150-151°.
C6H6O4Br2
Ber. C 23,80 H 2,0 Br 53,0
Gef. C 23,51 H 2,06 Br 52,55
Спектр IR. (KBr): группы и т.п.
при: 3430 (s), 3000 (s),
1710
(s), 1620 (w), 1405 (s), 1200 (s) см -1.
Спектр NMR. (CDCl3): δ = 2,33 (s/=/CHBr2),
7,75 (s/=/CH3) ppm.
(D-Ацетон)
2 фракцию: 0,1g = 10,7% от Smp. 138°. При анализе дважды
выкристаллизировались эфир/гексан: Smp.
150-151°.
C6H6O4Br2 Ber. C
23,81 H 1,99 Br 52,97
Gef. C 23,51 H 2,06 Br 52,55
Спектр IR.
(KBr): группы и т.п.
при: 3200 (b), 1725 (s), 1650
(w), 1230 (s), 1150
(m), см -1.
Спектр NMR.
(CDCl3):
δ = 4,59 (s/-CH2Br) ppm.
(D-ацетон)
б) Посредством серебра-p-тозилата и муравьиной кислоты
11,038g XIXc
смешивались в 500-мл-грушевидной колбе с 140 мл. с муравьиной кислотой (50 Mol-Aeq) и 9,3g серебром-p-тозулатом (1,3 Mol-Aeq.) при 20° 14Std.
под светлым магнитным воздействием. Затем в смесь прибавляют эфир/ацетон 1:2.
Фильтровали посредством стеклянного фильтра G4, выпаривали в около 50 мл в
сложном эфире уксуса и встряхивали в выходе натрий-хлоридной кислоте с водой со
льдом, высушивали натриумсульфатом и выпаривали. Остаток, желтое масло, сушился
3 Std. в высоком вакууме: 5,0
g. После хроматографии в 100g силицагеле с бензолом/эфиром, 1,80g бромлактоном XXIIa (33%) получали желтое масло,
которое использовалось для дальнейших реакций.
Проба при анализе еще раз хроматографировалась, после чего
субстанция кристаллизовалась. Из бензола/петроэфира выкристовализировались, при
Smp. 85-86.
C6H5O4Br
Ber. C 32,60 H 2,28 Br 36,61
Gef. C 32,52 H 2,28 Br 36,97
Спектр IR. (CHCl3): группы и т.п. при: 3210 (w), 3010 (w), 1765 (s), 1730 (s) см -1.
Спектр NMR. (CDCl3): δ = 4,81
(s, ghb последовательности t/-CH2Br),
5,21 (s, при последовательности t/CH2-O-) ppm.
Ди-(2-метоксикарбонил-винил)-сульфид
XXIVb
Выход 1 мл триэтиламина в 10 мл эфира удовлетворялся
сероводородом и переводился в 1g (12mMol) пропиоловой кислоты-сложного эфир матила. После 3 Std./0°
переводили в хлорид метилена и встряхивали с выходом гидрогена натрия -
карбонатом и водой со льдом. Выход метиленхлорида: 1,37g (53%)
ди-(2-метоксикарбонил-винил)-сульфид. В ходе анализа выкристализировался три
раза из метанол/эфира: Smp.
132.
C8H10O4S Ber. C 47,53 H 4,99 S 15,80
Gef. C 47,47 H 4,91 S 15,73
Спектр IR.
(CHCl3): группы и т.п. при: 2920 (w), 1700 (s), 1575 (s), 1430
(w), 1160 (s)
см -1.
UV. – спектр (C2H5OH) λmax 303
nm (ε =26300)
Спектр NMR. (CDCl3): δ = 3,78 (s/-O-CH3),
5,73/7,70 ABtrans (d/J= 12,5 Hz/-CH=CH-) ppm.
trans-β-бензилтио-акриловая
кислота-сложный эфир метила XXVb
800 мг 70-proz выхода натриумгидрогенсульфида (10 mMol NaHS) в 6 мл метанола переводились
при 0° под азотом с 840мг (12mMol)
сложным эфиром пропиоловой кислоты-метила. После 5Std. получали при охлаждении льдом 1,2 мл
(10 mMol) бензилбромида.
После дальнейших 40 минут заливался в метиленхлорид и встряхивался с выходом
натриумгидрогенкарбоната и водой со льдом. После высыхания и испарения
следовали 1,80g частично кристаллизовавшегося продукта, который представлял
собой смесь льда и транс-сложного эфира в отношении 3:7 согласно спектру NMR.
После фильтрации более 70g силицагелем мог кристаллизоваться из этого 925мг
(48%) trans-β-бензилтио-акриловая
кислота-сложный эфир метила; после двукратного выкристаллизирования сложного
эфира/эфира петрола, Smp.
66-67˚.
C11H12O2S Ber. C 63,45 H 5,81 S 15,4
Gef.
C 63,48 H 5,81 S 15,26
Спектр IR.
(CHCl3): группы и
т.п. при: 2990 (w), 2940 (w), 1690 (s), 1590 (s), 1440 (w), 1430 (m), 1160 (s), 950 (s)
см -1.
Спектр NMR. (CCl4): δ = 3,67 (s/-OCH3),
4,02(s/-CH2-S-), 5,86/7,90 ABtrans (d/J= 15,5
Hz/-CH=CH-), 7,39 (s/C6H5-) ppm.
trans-β-бензилтио-акриловая
кислота XXVa
Из ощелочения trans-β-бензилтио-акриловая
кислоты - сложного эфира метила 208 мг (1 mMol) выходило 3 мл спирта и нагревалось в 1,2 мл 1N щелочи питьевой соды 1/2 Std. в водяной бане. Держали в
эфире и встряхивали с выходом гидрогенкабонатом натрия. После заквашивания и
обратного встряхивания в метилхлолриде, высыхания последнего в сульфате натрия
и испарении получалось 120 мг (62%) кристаллизовавшейся транс-кислоты.
Выкристаллизация 3 раза из ацетона/гексана. Smp. 160-161°.
C10H10O2S Ber. C 61,85 H 5,19 S 16,51
Gef. C 71,69 H 5,05 S 16,17
Спектр IR. (CHCl3): группы и т.п. при: 3000
(b), 1705 (s), 1565 (s), 1430 (m), 1160 (s) см -1.
Спектр NMR. (CCl4): δ = 4,08(s/-CH2-S-),
5,86/7,90 ABtrans (d/J= 15,5 Hz/-CH=CH-), 7,39 (s/C6H5-)
ppm.
Cis-β-бензилтио-акриловая кислота XXVIa
372 мг (3mMol)
бензилмеркаптама, 3 мл метанола, 210 мг (3 mMol) пропиоловой кислоту и 1,5 мл 2N выхода натриумметилата находились при 0˚ под азотом с
магнитным воздействием 1/2 Std.
Затем опускались в хлорид метилена и встряхивались с выходом гидрогенкарбоната
натрия. выход гидрогенкарбоната натрия заквашивалась в 1N соляной кислоте и смешивался с
метиленхлоридом. После высыхания в сульфате натрия и испарения получали 574мг
(88%) кристаллического cis-β-бпензилтио-акриловой
кислоты, которая выкристаллизировалась 3 раза из ацетона/гексана: Smp. 138-139°.
C10H10O2S Ber. C 61,85 H 5,19 S 16,51
Gef. C 61,64 H 5,02 S 16,08
Спектр IR. (CHCl3): группы и т.п. при: 2930 (b), 1695 (s), 1565 (s), 1430 (m), 1160 (s) см -1.
Спектр NMR. (CDCl3/CCl4): δ = 4,0 (s/-CH2-S-),
5,89/7,25 ABcis (d/J= 10,5 Hz/-CH=CH-), 7,41 (s/C6H5-)
ppm.
Cis-β-бензилтио-акриловая
кислота-сложный эфир метила XXVII
100мг (1,52 mMol)
Cis-β-бензилтио-акриловой кислоты XXVIa мешались в эфире и
перемешивались с небольшим количеством диазометана. После около 5 минут суживались,
переливались в метиленхлорид и встряхивались с гидрокарбонатом натрия и водой
со льдом. После высыхания в сульфате натрия и выпаривании оставались 60 мг
бесцветного масла, согласно спектру NMR. cis-эфира.
C11H12O2S Ber. C 63,45 H 5,81 S 15,40
Gef. C 63,41 H 5,79 S 15,31
Спектр IR. группы и т.п.
при: 2990 (w), 2940 (w), 1690 (s)
(без растворителя) 1590 (s), 1440 (m), 1430 (w), 1360 (m), 1160 (s), 950 (s) см -1.
Спектр NMR. (CCl4): δ = 3,67 (s/-O-CH3),
3,90 (s/-CH2-S-), 5,77/7,02 ABcis (d/J= 10,5 Hz/-CH=CH-)
ppm.
Пропиоловая
кислота-t-сложный
эфир бутила XXIIIc
В смесь 10 мл пропиоловой кислоты (0,162 Mol), 25 мл диоксана и 1 мл серной
кислоты-моногидрата добавилистал в 100мл-грушевидной колбе с газовой трубкой
при -15˚ изобутилен, дополнив до объемов, например, около 1/3. После
запирания в камере 48 Std.
при 20˚. Затем выпаривался, добавлялся в эфир, выход эфира встряхивался с
гидрогенкарбонатом натрия и водой, выпаривалась и дистиллировался:
Sdp.:
95-105°/Torr; 10329g (47,5%)
C7H10O2
Ber. C 66,14 H 7,91
Gef. C 66,69 H 8,05
Спектр IR.(CHCl3) группы и т.п. при: 3290 (m), 2950 (m), 2105 (m), 1700 (s), 1370 (m), 1155 (s) см -1.
Спектр NMR.
(CDCl3): δ
= 1,53 (s/-C(CH3)3), 2,80 (s/=C-H) ppm.
Cis- + trans-β-бензилтио-акриловая кислота-сложный эфир бутила XXVIc + XXVc
В трехшейную колбу налили под азотной атмосферой при
комнатной температуре к выходу 630 мг (5 mMol) под давлением 6,8 мл 0,735N выхода пропиоловой кислоты-t-сложного эфира бутила (в
диметилформамиде) и 5,00 мл (5 mMol)
N-Аэтил-диизопропиламина.
После 15 Std. при 20˚
раствор охлаждался до 0° и добавили 0,6 мл (5 mMol) бензилбромида. После 1/2 Std. при 20˚ его добавили в сложный эфир уксуса и встряхивали с
водой со льдом. Получали после высыхания сульфате натрия и выпаривания 1,23g
желтого масла, которое фильтровалось 37g алокса с бензолом: получили 1,03g
(84%) β-бензилтио-акриловая
кислоты - сложного эфира бутила (смесь cis- и trans- эфира).
C14H18O2S Ber. C 67,18 H 7,25 S 12,8
Gef. C 67,08 H 7,29 S 12,77
Спектр IR. (CHCl3): группы и
т.п. при: 2960 (m), 1680 (s), 1580 (s), 1445 (m), 1380
(m), 1365 (s), 1305 (s), 1140 (s) см
-1.
Спектр NMR. (CDCl3): δ =1,48 (s/-C(CH3)3) 3,90 (s/-CH2-S-)/3,97 (s/
-CH2-S-), 5,69/6,92 ABcis (d/J = 10Hz/-CH=CH-), 5,72/7,55
ABtrans (d/J=15Hz, -CH=CH), 7,30 (s/C6H5-)
ppm.
К щелочи добавили 300 мг (1,2 mMol) t-сложного эфира бутила с 2 мл муравьиной кислоты, причем сразу
началась выкристаллизироваться транс-β-бензилтио-акриловая кислота. После около 1 Std получили нескольким
количеством гексана: 174 мг (74%). Таким образом, полученная кислота совпадала
во всех качествах с выше описанной транс-β-бензилтио-акриловая кислотой.
3-[2-(t-бутоксикарбонил)-винилтио-метил]-4-(2-бутен)олид
XXIX
11,5 мл, 0,433 выхода N-натриумгидрогенсульфида (5 mMol NaHS) опустили в диметилформамид под азотом
в трехвыходной колбе при -30˚ под магнитом медленно нагревали с 650мг (5mMol) пропиоловой
кислоты-t-сложного эфира бутила при комнатной температуре. После 17 Std. охлаждали снова до -30˚ и
смешивались постепенно с 0,95 мл (5 mMol) в аэтил-диизопропил-амине и в 1150 мг (5 mMol) α-карбокси-β-бромметил-
Δα,β
- бутенолид. После 15 минут при -25˚ получался выход 0,6 мл (10 mMol) уксуса льда. Добавляли в
сложный эфир уксуса и встряхивали с выходом гидрогенкарбоната натрия и
удовлетворяемого раствора поваренной соли. После высыхания в сульфате натрия и
испарении после вращения следовали 1,03g (81%) XXIX. При анализе
выкристаллизировалось три раза из эфира/гексана: Smp. 64°.
C12H16O4S
Ber. C 56,24 H 6,29 S 12,67
Gef. C 56,44 H 6,16 S 12,49
Спектр IR.
(CHCl3): группы и
т.п. при: 2980 (m), 1785 (s), 1750 (s), 1700 (s), 1645
(w), 1595 (s), 1365 (s), 1365 (s),
1155 (s), 1040 (s) см -1.
UV. – спектр (C2H5OH) λmax 273 nm (ε =11700)
Спектр NMR. (CDCl3): δ = 1,49 (s/-C(CH3)3),
3,8 (m/-CH2-S-), 4,87(m/-CH2O-),
6,1 (m/=<HCO-), 5,71/7,43 ABtrans (d/J= 15,5 Hz/-CH=CH-) ppm.
3-[2-(t-бутоксикарбонил)-винилтио-метил]-2-карбокси-4-(2-бутен)олид
XXVIII
Выше описанная попытка, которая вела к сульфиду XXIX, повторялась; тем не менее, удалось при
доработке указать на то, чтобы температура никогда не поднималась выше 0˚.
Получили таким образом 1,14g (76%) аморфный 3-[2-(t-бутоксикарбонил)-винилтио-метил]-2-карбокси-4-(2-бутен)олид
XXVIII.
Спектр IR. (CHCl3): группы и
т.п. при: 2920 (b), 1780 (s), 1745 (s), 1700 (s), 1580
(m), 1365 (s), 1245 (s), 1155 (s) см
-1.
UV. – спектр (C2H5OH) λmax 271 nm (ε =8200)
Спектр NMR.
(CDCl3): δ
= 1,49 (s/-C(CH3)3),
4,38 (m/-CH2-S-), 5,11 (m/O>CH2), 5,80/7,52 ABtrans(d/J 15
Hz/-CH=CH-) ppm.
(2-t-бутоксикарбонилвинил)-аллил-сульфид XXXIb
6,8 мл 0,735N
гидросульфида натрия (5 mMol NaHS)
в диметил-формамиде смешивали при -30˚ с 630мг (5 mMol) пропиоловой кислотой-t-сложным эфиром бутила.
После 24 Std. при 20°
охлаждали снова до -30° и смешивали с 0,45 мл (5 mMol) амилбромида. Добавляли после 48 Std. при 20° в эфире и встряхивали на расстоянии с диметилформамидом
5 раз с водой. После высыхания в сульфате натрия и выпаривания получали 969мг
масла, которое дистиллировалось при 83°/0,001 Torr: получали 916 мг (92%) бесцветного масла.
C10H16O2S Ber. C 59,98 H 8,05 S 16,01
Gef. C 60,11 H 7,90 S 16,32
Спектр IR. (CHCl3): группы и т.п. при: 2990 (m), 1700 (s), 1595 (s), 1375 (m), 1320 (s), 1155 (s), 985 см -1.
UV. – спектр (C2H5OH) λmax 278 nm (ε =13800)
Спектр NMR. (CDCl3): δ = 1,49 (s/-C(CH3)3),
3,45 (d/J = 6 Hz/-CH2-S-), 5,1-5,7
(Sh/CH2=CH-), 5,73/7,55 ABtrans (d/J = 15Hz/ -S-CH=CH-) ppm.
(2-карбоксивинил)-аллил-сульфид XXXIa
345 мг (1,73 mMol)
(2-t-бутоксикарбонилвинил)-аллил-сульфид
XXXIb смешивали с 2 мл
муравьиной кислоты 48 Std.
при 20˚. Затем добавляли в эфир и встряхивали с выходом гидрогенкарбонатом
натрия. После заквашивания и соединения с эфиром, высыхания и испарения
оставалось масло, которое дистиллировалось при 130°/0,001 Torr: оставалось 176 мг бесцветного масла
(71%).
C6H8O2S Ber. C 50,00 H 5,60 S 22,25
Gef. C 49,80 H 5,54 S 22,25
Спектр IR. (CHCl3): группы и т.п. при: 2980
(b), 1685 (s), 1580 (s), 1410 (m), 1265 (s), 985 (m) см -1.
UV. – спектр (C2H5OH) λmax 275 nm (ε =17000)
Спектр NMR. (CDCl3): δ = 3,49 (d/J = 6Hz/-CH2-S-),
5,1-6,3 (Sh/CH2=CH-), 5,79/7,78 ABtrans (d/J = 15
Hz/-S-CH=CH-) ppm.
(2-карбоксивинил)-(1-пропенил)-сульфид
XXXIIa
144 мг (1 mMol)
(2-карбоксивинил)-(1-пропенил)-сульфид XXXIIa нагревали в 5 мл диметилсульфоксида с 300 мг (0,26 mMol) калий-t-бутоксидом 2 Std. в масляной ванне на 90°. После
охлаждения остаток держали в эфире. После встряхивания с 1N соляной кислотой в воде со льдом и
выпаривания оставался коричневый остаток 105 мг. Из него кристаллизировали в
холодильнике в состояние 51мг. Изомеризированной кислоты XXXIIa. Кристаллизовали 3 раза в
эфире/гексане до. Smp. 109°.
C6H8O2S Ber. C 50,00 H 5,60
Gef. C 50,11 H 5,79
Спектр IR. (KBr): группы и
т.п. при: 3420
(b), 2990 (b), 1675 (s), 1565 (s), 1250 (s), 1185 (s), 950 (s), 805 (m), 775
(m) см -1.
UV. – спектр (C2H5OH) λmax 286 nm (ε =15800)
Спектр NMR. (CDCl3): δ = 1,88 (d/J = 5Hz/=/CH3), 6,11 (CH3-CH=CH-),
5,82/7,78 ABtrans
(d/J = 15 Hz/-CH=CH-CO-) ppm.
2-(метоксикарбониламино)-3-метил-4-(2-бутен)олид
XXXVIa
Образование промежуточных
ступеней стало следствием IR.
спектров
a) Хлорид кислоты XXXIII
710 мг (5 mMol)
α-карбокси-β-метил- Δα,β - бутенолид
смешивались с 15 мл метиленхлорида и с 1,2g фосфорпентахлорида (ber. 1,04g). После 30 минут при 20˚
встряхивали с водой со льдом. После высыхания в сульфате натрия и выпаривания
оставался остаток 794мг (согласно IR.
спектра хлорид кислоты).
б) ацид кислоты XXXIV
Вышеупомянутый хлорид кислоты, расширенный 10мл ацетона,
смешивался с 450мг (ber.
405мг) тонко распределенного калиумацида 1 Std. при 20°; затем охлаждался до 0°, добавлялся в 15мл
ледяного 50-proz. ацетона, 15 минут при 0°, выпаривался со сложным
эфиром уксуса: 375 мг (согласно спектру IR. смесь из ацида кислоты и кислоты).
Водянистая фаза заквашивалась и со сложным эфиром уксуса перемешивалась: 195 мг
исходного материала. 375 мг принимались с целью растворения кислоты в
метиленхлориде и встряхивались со сконцентрированным ледяным выходом
гидрогенкарбоната натрия. После высыхания и выпаривания остатка миетиленхлорида
оставалось 118 мг ацида кислоты. Выходом гидрогенкарбоната натрия стал сложный
эфир уксуса: 182 мг исходной субстанции.
в) изоцианат XXXV
118 мг ацида кислоты нагревалиь в 5 мл толуола 5 минут до
кипячения. Выпаривание в вакууме давало в итоге 105 мг (IR. спектр: изоцианат).
г) метилурэтан XXXVIa
Изоцианат нагревали с 3 мл метанола 5 минут на водяной бане.
После выпаривания оставалось 100мг метилурэтана. Чистое дистиллирование
проходит при 100°/0,001 Torr.
C7H9O4N Ber. C 49,12 H 5,30
Gef. C 48,92 H 5,30
Спектр IR. (CHCl3): группы и т.п. при: 3440 (w), 2940 (w), 1750 (b), 1690 (m), 1325 (m), 1040 (s) cм -1.
Спектр NMR. (CDCl3): δ = 2,19 (s/=CH3),
3,79 (s/-O-CH3), 4,74 (s/-O>CH2),
6,65(b/-NH) ppm.
Бензилурэтан XXXVIc, выкристаллизация из
Ацетон/гексана, Smp.
109-110˚
C14H13O4N Ber. C 63,16 H 5,26
N 5,67
Gef. C 63,15 H 5,30 N 5,67
Спектр IR. (CHCl3): группы и т.п. при: 3350 (m), 2940 (w), 1750 (b), 1690 (m), 1325 (s), 1040 (s) cм -1.
Спектр NMR. (CDCl3): δ = 2,12 (s/=/CH3),
4,67 (s/-O>CH2), 5,15 (s/-CH2-C6H5),
6,68(b/-NH), 7,36 (s/-C6H5) ppm.
Аэтилурэтан XXXVIb, выкристаллизация из
ацетона/гексана, Smp.
98-99˚
C8H11O4N Ber. C 51,88 H 5,99
N 7,56
Gef. C 51,90 H 5,92 N 7,60
Спектр IR. (CHCl3): группы и
т.п. при:
3400 (m), 2970 (w), 1780 (s), 1755 (s), 1690
(m), 1530 (m), 1325 (m), 1055 (s) cм -1.
Спектр NMR. (CDCl3): δ = 1.3 (t/J = 7Hz/CH3-CH2-),
2,17 (s/=/CH3), 4,22 (q/J = 7Hz/-CH2-O-), 4,72 (s/-O>
CH2), 6,63 (b/-NH) ppm.
ЗАКЛЮЧЕНИЕ
Дифенолы (резорцин, оливетол) могут смешиваться с α,
β - спиртами при посредничестве N, N-диметилформамид-динеопентилацетала.
Непосредственное образование от (-)-каннабидиола из (+) - trans или
(+)-cis-p-ментадиена-(2,8)-ol-(l) и оливетола открывает доступ к серии
оптически активных компонентов гашиша и определенно одновременно к их изучению.
Схема описывается бутенолидом типа XI, и обсуждаются опыты,
которые делались при конденсации этой связи с β-меркапто-акрил сложными
эфирами.
СПИСОК ЛИТЕРАТУРЫ
1) Zusammenfassende Arbeiten: a) A.R.Todd, Experimentia 2_, 55
(1946);b) D.F.Dowing, Quart. Rev. 16_, 133 (1962);
c) U.Clausen und F. Körte, "Die
Naturwissenschaften" 21, 541
(1966).
2)
T.Petrzilka, W.Haefliger, C.Sikemeier, G.Ohloff und A.
Eschenmoser, Helv. 50, 719 (1967).
3)
R.Adams, C.K.Cain, W.D.Mc Phee und R.B.Wearn, J. Amer. Chem.
Soc. 63, 2209 (1941).
4)
Y.Goani und R.Mechoulam, Tetrahedron 22, 1481 (1966).
5)
T. und H.Smith, J.chem. Soc. 21, 47 (1857).
6)
T.B. Wood, W.T.N.Spivey und T.H.Easterfield, J. chem. Soc.
75, 20 (1899).
7)
R.S.Cahn, J. chem. Soc. 1933, 1400.
8)
R.Adams, B. R. Baker und R.B.Wearn, J. Amer.
Chem. Soc. 62, 2204 (1940).
9)
R.Ghosh, A.R.Todd und
S.Wilkinson, J. chem. Soc. 1940, 1393.
10)
R.Adams, M.Hunt und J.H. Clark, J.
Amer. Chem. Soc. 62, 196 (1940).
11)
A.Jacob und A.R.Todd, Nature U5, 350 (1940) und J. chem. Soc. 1940
(649).
12)
F. Körte und H.Sieper, Liebigs Ann.
Chem. 630_, 71
(1960).
13)
R.Mechoulam und Y.Shvo, Tetrahedron 1£, 2073
(1963).
14)
F. Körte, E.Hackel und H.Sieper, Liebigs Ann. Chem. 685, 122 (1965).
15)
E.Dlugosch, U.Claussen und F.Körte, Liebigs Ann. Chem. 693, 165 (1966).
16)
R.Mechoulam und Y.Gaoni, J. Amer. Chem. Soc. 87_,
3273 (1965).
17)
R.Mechoulam und Y.Gaoni,
J. Amer. Chem. Soc. 86, 1646 (1964).
18)
R.L.Hively, W.A.Mosher und F.W.Hoffmann, J. Amer. Chem.
Soc.
88, 1832 (1966).
19)
E.C.Taylor, K.Lenard, Y.Shvo,
J. Amer. Chem. Soc. 88,
367 (1966).
20)
K. E . Fahrenholz, M.Lurie und R . W. Ki er stead , J. Amer. Chem. Soc. 88,
2079 (1966).
21)
A. W. D. Avison , A. L, Murrison und M.W.Parkes, J. ehem. Soc.
1949, 952.
22)
R.Adams
und T. E. Bocks tahle r , J. Amer. Chera. Soc. 74,
5346 (1952) und frühere Arbeiten.
23)
E.C.Taylor und E . J .
S tro j ny , J.
Amer. Chem. Soc. 82, 5198 (1960).
24)
E.C.Taylor,
K.Lenard und B.L.Loev, Tetrahedron 23_, 77 (1967).
25)
H. Budzikiewicz
et al., Tetrahedron 21_, 1881 (1965).
26)
U.Claussen und F.
Körte, Tetrahedron, Supplement No. 7, 89(1966).
27)
U.Claussen,
H. -W. Fehl habe r und F. Körte, Tetrahedron 22, 3535 (1966).
28)
P.B.Rüssel, A.R.Tood, S.Wilkinson, A.D.McDonald und G.Woolfe, J. chem. Soc. 1941, 169.
29)
R.Adams, M.Harfenist und S.Loewe, J. Amer. Chem. Soc.
71_, 1624
(1949).
30) H.
Gayer, Naunyn-Schmiedeberg's Arch.
exptl. Pathol. u.
Pharmakol. 129,
312 (1928).
31)
S.Fränkel,
Naunyn'-Schmiedeberg's Arch.
exptl. Pathol. u.
Pharmakol. 49, 266 (1903).
32)
C.S. Parker und
F.Wringley, J. Ment. Sei.
96, 276 (1950).
33)
J.L.Simonsen und A.R.Todd, J. chem. Soc. 1942, 188.
34) J.Levtne, J. Amer. Chem. Soc. 66,
1868 (1944).
35) Z.Krejci und F.Santavy, Acta Univ. Palackianea Olomuc. 6_, 59 (1955).
36)
O.E.Schultz und G.Haffner, Arch.
Pharm. 293/65, 1
(1960).
37)
W. v. E. Doering und W.R.Roth, Telrahedron 18, 67(1962).
38)
R.K.Hill
und
A.G.Edwards,
Tetrahedron Letters 1964,
3239.
39)
R.F.Church und R.E.Ireland, J. org. Chem. 2jB,
17(1963).
40)
A. W. Burgstahler
und J.C.
Nordin, J. Amer. Chem. Soc. 83, 198 (1961).
41)
H. Brechbühler, H.Büchi, E.Hatz, J.Schreiber und A. Eschenmoser, Helv.
48, 1746 (1965).
42)
G.O.Schenck, K.Gollnick, G.
Buchwald, S. Schröter und G.Ohloff, Liebigs Ann. Chem. 674,
93 (1964).
43)
G.Ohloff, K. W. Schulto-Eltc und W.Giersch, Helv.
4£, 1G65
(1965).
44)
K.Gollnick und
G.Schade, Tetraheclron Letters
1966, 2335.
45)
C.M.Suter und A.W.Weston, J. Amer. Chem. Soc.
61, 232(1939).
46)
L. M. Jackmnn,
NMR Spcclroscopy in Organic Chemistry,
(Pergamon Press, New York,
1959, S. 125).
47)
H.B.Henbest und
R.S. Mc Elhinney,
J. chem. Soc. 1959, 1834.
48)
Y.-R.Naves
und
A. V. Grampoloff, Bull. Soc.
chim. France 1960, 37.
49)
A. J.Birch, Annu.
Rep. Progr. Chem. 47,
191 (1950).
50)
R.S.Cahn, CK.Ingold
& V.Prelog, Experimentia 12, 81(1956).
51)
R.Adams,
M.Hunt und J.H. Clark, J. Amer. chem. Soc. 62,735 (1940).
52)
N. I. Kursanov,
J. Russ. Phys.
Chem. Soc. 46,
815 (1914).
53)
R.Mechoulam
und Y.Gaoni, Tetrahedron Letters 12, 1109 (1967).
54)
A.D.Cross,
Introduction to Practical Infra-Red
Spectroscopy (Butterworth Publ. 1960).
55)
W.Haefliger und T.Petrzilka, Helv. 49,
1937 (1966).
56a) G. G. F. Newton und E.P.Abraham, Nature 175,
548(1965). b) G.G. F.
Newton und E.P.Abraham, Biochem.
J. 62, 651
(1956).
57)
E.P.Abraham und G. G.
F. Newton, Biochem. J.
79, 377(1961).
58)
D.C.Hodgkin und E.N.Maslen,
Biochem. J. 79_,
393 (1961).
59)
B.Loder, G.G. F.
Newton und E.P.Abraham, Biochem. J.
79,
408 (1961).
60)
R. R. Chauvette
et al., J. Amer.
Chem. Soc. 84,
3401 (1962).
61)
S.Eardley und A.G.Long, Deutsches Patent Nr. 1228 264.
62)
D.M.Green,
A.G.Long, P. J.
May und A.F.Turner, J. ehem. Soc.
766, (1964).
63)
G.C.Barrett,
S.H.Eggers, T.R.Emerson und G.Lowe, J.
chem. Soc. 788, (1964).
64)
E.Galantay,
H.Engel, A.
Szabo und J.Fried, J. Amer.
Chera. Soc. 3360,
29 (1964).
65)
J.C.Sheehan
und
D.A.Johnson, J. Amer.
Chem. Soc. 76,
158 (1954).
66)
R.B.Morin et
al., J. Amer. Chem. Soc. 85_,
1896 (1963).
67)
R. B. Woodward, Angew. Chem.
78, 557 (1966).
68)
R.B. Woodward,
K. Heusler, J.Gosteli, P. Naegeli, W.Oppolzer, R.Raraage, S . Ranganathan, H . Vorbrüggen, J. Amer. Chem. Soc. 88,
852 (1966).
69)
R.Graf, Angew.
Chem. 74, 523 (1962).
70)
A.C.Cope et
al., J. Araer. Chem. Soc.
60, 2644 (1938).
71)
J. Kollonitsch, A.Rsegay und G.Doldovras, J. Amer. Chem. Soc. 86, 1857 (1964).
72)
W.E.Truce, L.N.Owen
und
M. U . S . Sultanbn wa , J. Amer. Chem. Soc.
83, 4636 (1961), und frühere
Arbeiten.
73)
F.Montanari und A.Negrini, Chem.
Zbl. 1959, 2797,
und frühere Arbeiten.
74)
F. Bohlmann und E.Bresinsky, Chem.
Ber. 96, 584 (1964); 97, 2109 (1965).
75)
A.A.Oswald et al.,
J. Amer. Chem.
Soc. 86, 2876 (1964).
76)
E. Winterfeld und H.Preuss,
Chem. Ber. 98,
3537 (1965); 99, 450
(1966).
77)
M. Widmer, Dissertation ETH, Zürich 1962.
78)
D.S. Tarbeil
und W.E.Lovett, J. Amer. Chem. Soc. 78,
2259 (1956).
79)
D.Martin, A.Weise und H.-J.Niclas, Angew. Chem. 79,
340 (1967).
80) C.Pascual, J.Meier
und
W.Simon, Helv. 49.
164(1966).
АВТОБИОГРАФИЯ
Я родился 14 декабря 1939 года в Лангнау. После посещения
начальной школы в Лангнау и средней школы в Сюррее, я поступил в школу кантона
Люцерн, где я сдал осенью 1959 года экзамен на аттестат зрелости тип C. В том
же году я начал мое обучение в относящемся к клятвенному союзу техническом
институте и получил весной 1964 года диплом как естественно-научный специалист
(химически-физическое направление). С тех пор я работал в группе профессора
доктора А. Эшенмосера под руководством доктора Т. Петрцилка над данной работой
для получения ученой степени кандидата наук.
Цюрих, июнь 1967 года
Вальтер Хэфлигер