МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
Харьковский национальный университет
им. В.Н. Каразина
“К ЗАЩИТЕ”
Заведующий кафедры материалов реакторостроения
Проф*******
|
ВЫПУСКНАЯ РАБОТА
МАГИСТРА ПРИКЛАДНОЙ ФИЗИКИ
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ПЛАСТИЧЕСКОЙ ДЕФОРМАЦИИ И РАЗРУШЕНИЯ ГПУ КРИСТАЛЛОВ
| Руководитель |
************
|
| Студент |
************** |
Харьков
2008
Содержание
| Введение |
| 1 |
Применения МД для исследования пластической деформации кристаллов |
| 1.1 |
Ограничения |
| 1.2 |
Потенциал |
| 1.3 |
Алгоритм интегрирования по времени |
| 1.4 |
Процедура минимизации |
| 1.5 |
Вычисление сил |
| 1.6 |
Периодичность |
| 1.7 |
Начальное состояние |
| 1.8 |
Начальное состояние для кристалла с дефектами |
| 1.9 |
Нагрузка |
| 1.10 |
Уравнение для ширины ячейки моделирования |
| 1.11 |
Контроль системы |
| 1.12 |
Вычисление физических величин |
| 1.13 |
Визуализация |
| 2 |
Моделирования пластической деформации ГПУ кристаллов |
| Заключение |
| Список использованных источников |
Анотація
В даній роботі проведено аналіз особливостей застосування методу молекулярної динаміки для моделювання пластичності кристалів. Запропоновано новий підхід до моделювання розтягуваннякристалів. Запропоновано динамічне рівняння для поперечного розміру коміркимоделювання. Створена програма для дослідження процесу пластичної деформації та руйнування кристалів. Проведено моделювання розвитку пластичної деформації ГЩУ кристалів при одноосному розтягуванні. Показана принципова можливість імітації за допомогою цього методу кривих розтягуваннядосконалихкристалів, зміни температури зразка, появи дислокацій, полос ковзання, поодиноких вакансій та їх скупчень, а також процесу руйнування кристалів
Аннотация
В данной работе проведен анализ особенностей применения метода молекулярной динамики для моделирования пластичности кристаллов. Предложен новый подход к моделированию растяжения кристаллов. Предложено динамическое уравнение для поперечного размера ячейки моделирования. Создана программа для исследования процесса пластической деформации и разрушения кристаллов. Проведено моделирование развития пластической деформации ГПУ кристаллов при одноосном растяжении. Показана принципиальная возможность имитации с помощью этого метода кривых растяжения совершенных кристаллов, изменения температуры образца, появления дислокаций, полос скольжения, одиночных вакансий и их скоплений, а также процесса разрушения кристаллов.
Abstract
In the given article the analysis of features of application of a molecular dynamics for simulation of a plasticity of crystals is conducted. The new approach to simulation of strain of crystals is offered. The dynamic equation for transverse dimensions of simulation cell is offered. The code for investigation of the process of a plastic deformation and destruction of crystals is created. The simulation of development of a plastic deformation hcp crystals is carried out at monoaxial expansion. The principal possibility of imitation with the help of this method of stress-strain curves for the perfect crystals, temperature variation of a sample, appearance of dislocations, stripes of slide, single vacancies and their clusters, and also process of crystal destruction.
Введение
Бурный рост ядерной энергетики и широкое развитие работ в области термоядерного синтеза послужили мощным стимулом интенсификации исследований в области облучаемых материалов. Создана новая ветвь материаловедения – атомное, радиационное материаловедение.
Свойства материалов всегда были ключевым звеном, определяющим успех многих инженерных разработок в различных областях техники. Особенно их роль возросла в последнее время при создании сложных конструкций, работающих в экстремальных условиях. Ядерные реакторы, устройства термоядерного синтеза – яркий пример таких конструкций. Сотни различных по составу, структуре и способам изготовления материалов обеспечивают их работоспособность. Но, попадая в условия высоких потоков облучения (до 1020
нейтр/м2
∙с) и больших флюенсов (до1027
-1028
нейтр/м2
), они претерпевают значительные структурные перестройки (радиационное повреждение). Следствием этих перестроек является резкое изменение всех физических свойств материалов. Причем эти изменения носят не совсем обычный характер. Ранее ничего подобного не встречалось в обширной практике работ с различными материалами. Так, были обнаружены абсолютно новые явления, происходящие с облученными материалами и сплавами: радиационное охрупчивание, радиационное распухание, радиационное упрочнение, ускоренная диффузия, радиационно-индуцированные фазово-структурные превращения и др.
И этот перечень, судя по всему, будет продолжен, так как исследования в области термоядерного синтеза уже выдвигают новые требования и к конструкционным материалам, которые должны будут работать в еще более жестких условиях (например, выдерживать облучение нейтронами с энергией до 14 МэВ).
Одним из основных факторов, определяющих свойства материалов, как известно, является структура. В условиях облучения она претерпевает существенные перестройки на атомарном уровне. Чтобы выявить принципиальные закономерности поведения материалов в тех или иных условиях эксплуатации, создать материалы с заданными свойствами, необходимо установить связь между изменяющейся атомарной структурой материалов и всей совокупностью их макроскопических свойств. Для решения этой важной задачи радиационного материаловедения привлекаются самые современные физические методы анализа структуры материалов, начиная с уже ставшего традиционным рентгеноструктурного анализа и кончая автоионный микроскопией, оже-спектроскопией и др.
Но в значительном числе случаев все же не удается произвести необходимые структурные изменения. Такая ситуация возможна при известных, но быстро меняющихся внешних условиях (например, ударное воздействие на материал при пролете высокоэнергетических нейтронов). Если же точно не известны изменения в структуре материала на всех этапах какого-либо технологического процесса, то трудно говорить о каком-либо надежном прогнозе его поведения.
В ряде случаев современная аппаратура из-за своего недостаточного разрешения не позволяет наблюдать атомные перестройки в материалах, например отдельные атомные скачки при диффузии, растворении и рост предвыделений второй фазы в сплавах и т.д.
Чтобы преодолеть перечисленные трудности и воссоздать быстроразвивающиеся процессы в материалах, перестройки на атомарном уровне или процессы, когда доступ к материалам ограничен или опасен, все чаще в атомном материаловедении привлекается компьютерный эксперимент. Обзор методов компьютерного моделирования используемых в радиационном материаловедении на начало 90-х годов можно найти в [1].
Метод молекулярной динамики (МД) - один из наиболее мощных методов, используемых для компьютерного моделирования в радиационном материаловедении. Он позволяет проводить детальные исследования структуры материалов исходя из первых принципов. В последние годы метод молекулярной динамики переживает второе рождение. Связано это, во-первых, с быстрым увеличением мощности компьютеров (быстродействия, объема памяти), что позволяет проводить беспрецедентные по масштабам компьютерные эксперименты. Так, сегодня уже проведены расчеты пластической деформации нанокристаллов меди, состоящих из 100 миллионов атомов [2]. С другой стороны, были разработаны более совершенные потенциалы межатомного взаимодействия, включающие в себя также многочастичные эффекты.
Данная работа является продолжением защищенной в 2003 г. бакалаврской работы, в которой были проанализировано современное состояние методов математической обработки кривых растяжения реакторных материалов, а также было проведено, на примере реакторных сталей, определение основных параметров, характеризующих пластическую деформацию.
Целью работы является изучение метода молекулярной динамики и особенностей его применения к исследованию пластичности реакторных материалов, разработка алгоритма и создание программы для изучения пластичности двумерных ГПУ-кристаллов.
1. Применения МД для исследования пластической деформации кристаллов
Для изучения пластических свойств материалов методом МД, атомы из которых состоит материал, помещают в ячейку моделирования, обычно прямоугольной формы, которую подвергают деформации. Координаты и скорости атомов находят, численно решая уравнения движения Ньютона для заданных потенциалов взаимодействия между атомами. Затем, зная координаты и скорости атомов, с помощью соответствующиго усреднения, вычисляют макроскопические величины, такие как температура, давление, тензор напряжения и т.д. Достоинство метода состоит в возможности получить физические величины из первых принципов, т.е. используя только уравнения движения и потенциал межатомного взаимодействия.
1.1. Ограничения
Укажем на ограничения, свойственные этому методу. Во-первых, можно сразу же спросить: почему мы используем законы Ньютона, чтобы двигать атомы, хотя известно, что системы на атомном уровне подчиняются скорее квантовым законам, чем классическим?
Простейшая проверка применимости классического приближения базируется на тепловой длине волны де-Бройля, определяемой как
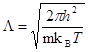 , , |
(1) |
где 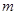 - атомная масса и - атомная масса и  - температура. Классическое приближение хорошо работает когда - температура. Классическое приближение хорошо работает когда  , где , где 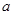 - среднее расстояние между атомами. Если рассматривать, например, жидкость в тройной точке, то - среднее расстояние между атомами. Если рассматривать, например, жидкость в тройной точке, то 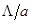 порядка 0.1 для легких элементов, таких как Li и Ar, уменьшаясь для более тяжелых элементов. Классическое приближение плохо работает для легких систем, таких как H2
, He, Ne. порядка 0.1 для легких элементов, таких как Li и Ar, уменьшаясь для более тяжелых элементов. Классическое приближение плохо работает для легких систем, таких как H2
, He, Ne.
Кроме того, квантовые эффекты становятся важными для любых систем, когда температура достаточно низка. Падение удельной теплоемкости кристаллов ниже температуры Дебая и аномальное поведение коэффициента теплового расширения есть хорошо известные примеры измеримых квантовых эффектов в твердых телах. Поэтому результаты, полученные с помощью МД должны интерпретироваться с известной осторожностью в этих областях.
Второе ограничение связано с ограниченностью используемых компьютерных ресурсов, что приводит к ограничению количества рассматриваемых атомов и, как следствие, к снижению точности вычисляемых физических величин. Частично эту проблему можно обойти, используя подходящие граничные условия (см. ниже). Постоянный рост мощности компьютеров также способствует смягчению этой проблемы. Так, в настоящее время, в печати имеются сообщения о расчетах с 100 млн. атомов.
Необходимо отметить, что современные мощные суперкомпьютеры являются параллельными. Поэтому для расчетов на них необходимо обеспечить эффективное распараллеливание вычислений. Об одной новой возможности распараллеливания в рассматриваемой здесь задаче будет сказано ниже.
Ограниченное быстродействие компьютеров накладывает ограничения на скорость деформации, используемую в вычислениях. Это связано с тем, что при решении уравнений движения шаг по времени должен составлять порядка 0,01 шага от периода колебаний атомов (по порядку величины равного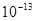 сек) для обеспечения необходимой точности вычислений. Отсюда следует, что для обеспечения деформации ~100% за приемлемое время вычислений типичная скорость деформации должна составлять порядка сек) для обеспечения необходимой точности вычислений. Отсюда следует, что для обеспечения деформации ~100% за приемлемое время вычислений типичная скорость деформации должна составлять порядка  сек сек в то время как максимально достигаемая в эксперименте скорость деформации составляет 105
сек-1
. Кроме очевидной возможности достижения более низкой скорости деформации, увеличивая время вычислений, есть еще одна возможность – использовать процедуру минимизации. в то время как максимально достигаемая в эксперименте скорость деформации составляет 105
сек-1
. Кроме очевидной возможности достижения более низкой скорости деформации, увеличивая время вычислений, есть еще одна возможность – использовать процедуру минимизации.
При нулевой температуре система находится в локальном минимуме внутренней энергии, а из-за отсутствия тепловых колебаний она не может покинуть этот минимум.
Процедура минимизации позволяет деформируемой системе находиться вблизи локального минимума внутренней энергии. При таком моделировании время не определено, так как мы не решаем уравнений движения. Следовательно, скорость деформации не оказывает никакого влияния, если она достаточно низка чтобы исключить нагрев системы, и обеспечить сходимость процедуры минимизации. Таким образом, моделирование, основанное на процедуре минимизации, представляет собой модель идеализированного эксперимента при нулевой температуре в пределе низкой скорости деформации, когда тепло выделяемое при деформировании, удаляется.
1.2. Потенциал
Для моделирования материала необходимо задать потенциал взаимодействия составляющих его атомов. Наиболее простым является парный потенциал Леннарда-Джонса
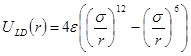 , , |
(2) |
Здесь, 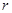 - расстояние между атомами, - расстояние между атомами, 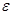 - глубина потенциальной ямы и - глубина потенциальной ямы и 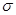 связано с положением минимума потенциала связано с положением минимума потенциала 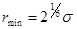 . Потенциал Леннарда-Джонса качественно правильно описывает взаимодействие между атомами – сильное отталкивание на малых расстояниях, обусловленное первым слагаемым в скобках, и притяжение на больших расстояниях, за которое отвечает второе слагаемое в скобках. Он хорошо описывает ван-дер-ваальсовское взаимодействие между атомами кристаллов благородных газов, но, вследствие своей простоты, часто используется для качественного описания взаимодействия других атомов. С потенциалом Леннарда-Джонса проведено большое количество вычислений. Он является стандартным в вычислениях методом МД. . Потенциал Леннарда-Джонса качественно правильно описывает взаимодействие между атомами – сильное отталкивание на малых расстояниях, обусловленное первым слагаемым в скобках, и притяжение на больших расстояниях, за которое отвечает второе слагаемое в скобках. Он хорошо описывает ван-дер-ваальсовское взаимодействие между атомами кристаллов благородных газов, но, вследствие своей простоты, часто используется для качественного описания взаимодействия других атомов. С потенциалом Леннарда-Джонса проведено большое количество вычислений. Он является стандартным в вычислениях методом МД.
Основными материалами реакторостроения являются металлы – сталь, цирконий и т.д. В металлах природа сил взаимодействия между атомами не двухчастичная (парная) а многочастичная. Effective Medium Theory (EMT) дает реалистическое описание металлической связи с учетом её многочастичной природы [3,4]. EMT- потенциал, с вычислительной точки зрения, не намного сложнее парного потенциала, но дает намного более реалистическое описание свойств материалов. Поскольку в данной работе не ставится задача изучения пластических свойств конкретного материала мы будем использовать модельный потенциал Леннарда-Джонса.
Удобно при этом выбрать в качестве единицы длины , единицы энергии и единицы массы - массу атомов (полагаем, что материал состоит из атомов одного сорта). Это приводит к ускорению вычислений. Чтобы перейти к величинам, характеризующим конкретный материал, необходимо ввести соответствующие масштабные множители - для длины, 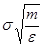 для времени, для времени, 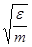 для скорости, для скорости, 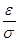 для силы, для силы, 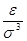 ( (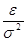 в двумерном случае) для напряжения, где в двумерном случае) для напряжения, где 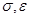 и взяты для данного материала. и взяты для данного материала.
Потенциал Леннарда-Джонса простирается до бесконечности. Однако на больших расстояниях он мал. И поэтому его влияние на движение далеких атомов мало. Чтобы ускорить вычисления эту несущественную часть потенциала отбрасывают, или, другими словами, вводят обрезание потенциала
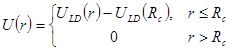 . . |
(3) |
Радиус обрезания 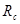 традиционно выбирают традиционно выбирают 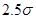 или или 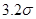 . Возможные причины такого выбора будут обсуждаться ниже. В данной работе будет использоваться радиус обрезания . Возможные причины такого выбора будут обсуждаться ниже. В данной работе будет использоваться радиус обрезания 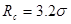 . Другие необходимые изменения в потенциале Леннарда-Джонса будут обсуждаться в разделе, посвященном выполнению закона сохранения энергии в МД. . Другие необходимые изменения в потенциале Леннарда-Джонса будут обсуждаться в разделе, посвященном выполнению закона сохранения энергии в МД.
1.3. Алгоритм интегрирования по времени
Основным компонентом программ, использующих метод молекулярной динамики, является алгоритм интегрирования по времени. Он необходим, чтобы проинтегрировать уравнения движения взаимодействующих частиц и найти их траектории.
Алгоритм интегрирования по времени основывается на методе конечных разностей, время при этом задается на конечной сетке, шаг по времени есть расстояние между последовательными точками сетки. Зная положения и скорости в момент времени 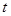 (точные детали зависят от типа алгоритма) схема интегрирования дает те же величины в более поздний момент времени (точные детали зависят от типа алгоритма) схема интегрирования дает те же величины в более поздний момент времени  . Используя процедуру интегрирования временную эволюцию системы можно прослеживать в течении длительного времени. . Используя процедуру интегрирования временную эволюцию системы можно прослеживать в течении длительного времени.
Конечно, эти схемы приближенными, и, поэтому, существуют ошибки, связанные с ними. Они классифицируются так:
Ошибки обрывания, связанные с точностью метода конечных разностей по отношению к истинному решению. Метод конечных разностей обычно базируется на ряде Тейлора, оборванном на некотором члене. Эти ошибки не зависят от программной реализации метода, они присущи самому алгоритму.
Ошибки округления, связаны с ошибками, возникающими при программной реализации алгоритма. Например, такие ошибки возникают из-за конечного числа цифр, используемых в компьютерной арифметике.
Оба типа ошибок можно уменьшить, уменьшая 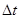 . Для больших ошибки обрывания доминируют, но они быстро уменьшаются, когда уменьшается. Например, алгоритм Верле имеет ошибки обрывания пропорциональные . Для больших ошибки обрывания доминируют, но они быстро уменьшаются, когда уменьшается. Например, алгоритм Верле имеет ошибки обрывания пропорциональные 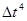 для каждого временного шага интегрирования. Ошибки округления падают более медленно с уменьшением и доминируют в пределе малых . Использование 64-битной точности (соответствующую “двойной точности” в Fortrane) помогает сохранить ошибки округления минимальными. для каждого временного шага интегрирования. Ошибки округления падают более медленно с уменьшением и доминируют в пределе малых . Использование 64-битной точности (соответствующую “двойной точности” в Fortrane) помогает сохранить ошибки округления минимальными.
В молекулярной динамике наиболее часто используемым алгоритмом интегрирования по времени является, вероятно, так называемый алгоритм Верле [5]. Основная идея состоит в том, чтобы записать разложение Тейлора до третьего порядка вперед и назад по времени. Пусть 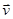 обозначает скорость, обозначает скорость, 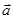 - ускорение и - ускорение и  - третью производную от - третью производную от 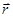 по . Тогда имеем: по . Тогда имеем:

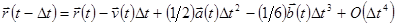 . .
|
(4) |
Складывая эти 2 выражения получаем
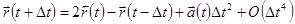 . . |
(5) |
Это основная формула алгоритма Верле. Так как мы интегрируем уравнения Ньютона, то  есть просто сила, деленная на массу, и сила в свою очередь есть функция положения есть просто сила, деленная на массу, и сила в свою очередь есть функция положения  : :
 . . |
(6) |
Видно, что ошибка обрывания алгоритма, когда система эволюционирует в течении времени , есть величина порядка , даже если третья производная не появляется в явном виде. Этот алгоритм в тоже время является простым для программной реализации, точным и стабильным, что объясняет его большую популярность при МД моделировании.
Проблема с этой версией алгоритма Верле состоит в том, что скорости явно не вычисляются. Хотя они не нужны для временной эволюции, но их знание иногда необходимо. Кроме того, они нужны для вычисления кинетической энергии 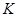 , чья оценка необходима чтобы проверить сохранение полной энергии , чья оценка необходима чтобы проверить сохранение полной энергии 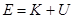 . Это один из наиболее важных тестов, указывающих, что МД моделирование выполняется корректно. Можно вычислить скорости из положений использую формулу . Это один из наиболее важных тестов, указывающих, что МД моделирование выполняется корректно. Можно вычислить скорости из положений использую формулу
 . . |
(7) |
Однако, ошибки, которые дает это выражение, порядка  а не . а не .
Чтобы преодолеть эту трудность, были развиты варианты алгоритма Верле. Они дают точно ту же траекторию и отличаются переменными, которые хранятся в памяти. Leap-frog алгоритм есть один из таких вариантов.
Лучший реализацией того же основного алгоритма есть так называемый алгоритм Верле со скоростью, когда положение скорости и ускорения в момент времени получается из тех же величин в момент времени следующим образом:
Заметим, что необходимо 9N ячеек памяти, чтобы сохранить 3N положений, скоростей и ускорений, но нам не нужно одновременно хранить значения любой из этих величин для двух различных времен.
1.4. Процедура минимизации
Чтобы моделировать деформацию при нулевой температуре используется процедура минимизации, которая позволяет поддерживать систему вблизи локального минимума энергии все время. Деформация и минимизация выполняются одновременно. Алгоритм минимизации представляет собой модифицированный алгоритм МД. После каждого шага по времени МД для каждого атома вычисляется скалярное произведение между импульсом и силой. Для атомов, скалярное произведение для которых отрицательно, импульс зануляется, так как эти атомы движутся в направлении, в котором потенциальная энергия возрастает. Таким образом, кинетическая энергия атомов удаляется, тогда как потенциальная энергия приближается к локальному минимуму энергии вдоль направления движения атома. Такая процедура минимизации быстро сдвигает систему в окрестность локального минимума энергии, но полной сходимости не получается, так как полная сходимость требует числа шагов по времени порядка числа степеней свободы системы. Однако, обычно увеличении числа шагов процедуры минимизации приводит лишь к малым изменениям в эволюции системы.
1.5. Вычисление сил
Наибольших вычислительных усилий требует вычисление сил, действующих между атомами. Поэтому оптимизации алгоритма вычисления сил необходимо уделить особое внимание. Один из шагов в этом направлении состоит в замене сложных для вычисления выражений для сил (например, содержащих экспоненту) на легко вычисляемые выражения (например, сплайны третьего порядка). Второй шаг состоит в использовании потенциалов с ограниченным радиусом действия, или, как указывалось выше, в обрезании несущественной области потенциала, если радиус действия потенциала бесконечен. При этом необходимо вычислить только силы, действующие со стороны ближайших атомов, т.е. находящихся внутри сферы (окружности в двумерном случае) с радиусом равным радиусу обрезания .
Третий шаг состоит в оптимизации алгоритма поиска атомов, ближайших к данному атому. Дело в том, что прямолинейный перебор всех атомов, вычисление расстояний до них и отбрасывание тех атомов, расстояние до которых превышает радиус обрезания , требует количества операций пропорционального 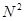 , где , где  - число атомов в системе. Следовательно, с ростом число требуемых операций быстро возрастает, и поэтому выполнение вычислений сильно замедляется, а, для больших , делается практически невыполнимым. Таким образом, чтобы избежать этого замедления нужен алгоритм, для которого число требуемых операций росло бы с линейно, а не квадратично. В принципе такой алгоритм прост – нужно перебирать не все атомы, а только достаточно близкорасположенные. Такое утверждение представляет собой тавтологию, пока не конкретизировано понятие близкорасположенных атомов. Чтобы сделать это, разобьем ячейку моделирования на более мелкие субячейки. Тогда близкорасположенные к данному атому будут атомы, которые расположены в субячейках, соседних с субячейкой, содержащей данный атом или в субячейках соседних с соседними. - число атомов в системе. Следовательно, с ростом число требуемых операций быстро возрастает, и поэтому выполнение вычислений сильно замедляется, а, для больших , делается практически невыполнимым. Таким образом, чтобы избежать этого замедления нужен алгоритм, для которого число требуемых операций росло бы с линейно, а не квадратично. В принципе такой алгоритм прост – нужно перебирать не все атомы, а только достаточно близкорасположенные. Такое утверждение представляет собой тавтологию, пока не конкретизировано понятие близкорасположенных атомов. Чтобы сделать это, разобьем ячейку моделирования на более мелкие субячейки. Тогда близкорасположенные к данному атому будут атомы, которые расположены в субячейках, соседних с субячейкой, содержащей данный атом или в субячейках соседних с соседними.
Удобно разбить ячейку моделирования на субячейки – параллелепипеды (прямоугольники в двумерном случае). Вследствие сильного отталкивания на малых расстояниях, атомы не могут подходить близко друг к другу. Поэтому можно выбрать такие размеры субячеек, что в каждой из них будет находится не более одного атома.
Таким образом, алгоритм поиска атомов, удаленных от данного атома на расстояние не больше радиуса обрезания , выглядит следующим образом. По номеру атома находим координаты атома и по ним субячейку, в которой находится атом. Затем находим субячейки, удаленные от нее на расстояние не более чем . Атомы, расположенные в этих субячейках, и будут искомыми (см. рис.1). Чтобы найти номер атома, хранящегося в заданной субячейке, удобно ввести массив, каждый элемент которого соответствует определенной субячейке. В этом элементе массива будет хранится номер атома, расположенного в этой субячейке, или нуль, если субячейка пуста. Элементы этого массива обновляются на каждом шаге по времени МД. Ясно, что изложенный алгоритм обеспечивает линейный рост числа операций с ростом числа атомов в системе. Вариации этого алгоритма используются в программах МД “Gromex”[6], “MOLDY”[7], “DL_POLY”[8] и др.
Возможна и другая организация вычислений, которая будет удобна для организации параллельных вычислений. Именно для вычисления сил, действующих на данный атом, можно перейти от суммирования по близлежащим атомам, к суммированию по близлежащим субячейкам (см рис.1). Будем двигаться последовательно по субячейкам первого ряда. Дойдя до конца первого ряда, перейдем в начало второго ряда и т.д.
| 1 |
2 |
3 |
4 |
| 5 |
6 |
7 |
8 |
| 9 |
10 |
11 |
12 |
Рис.1 Схема поиска ближайших атомов.
Если в субячейке находится атом, то вычисляем силу, действующую на него, со стороны ближайших атомов, расположенных в близлежащих субячейках. Если же субячейка пуста, то переходим к следующей. Отметим при этом, что, например, для атома находящегося в субячейке 6 (см. рис.1) необходимо вычислить силу, действующую со стороны атомов расположенных в субячейках 1, 2, 3, 7. Силы, действующие со стороны атомов, расположенных в субячейках 5, 9, 10, 11 в силу третьего закона Ньютона, с точностью до знака уже известны. Они были вычислены, когда вычислялись силы, действующие на атомы, расположенные в этих субячейках. Таким образом, в данной организации вычислений, необходимо рассматривать лишь половину близлежайших субячеек. Далее, при переходе к смежной субячейке 7 нет необходимости исследовать все близлежащие субячейки для поиска находящихся в них близко расположенных атомов. Необходимо лишь исследовать ячейки 4 и 8. И к найденным в них атомам, добавить атомы, найденные для ячейки 6, за исключением атомов находящихся в субячейках 1 и 6. Таким образом, информация о ближайших атомах для данной субячейки не теряется, а используется при поиске ближайших атомов для смежной субячейки. Это естественно приводит к ускорению вычислений.
1.6. Периодичность
Число атомов, помещенных в ячейку моделирования, намного меньше числа атомов входящих в состав макроскопических систем. Чтобы результаты нашего моделирования можно было распространить на макроскопические тела, делают допущение, что макроскопические системы, состоят из бесконечного числа периодически повторяющихся ячеек моделирования. Такая периодичность может быть в одном, двух и трех направлениях в трехмерном случае и в одном и двух в двумерном случае (см. рис.2). В этой работе мы будем рассматривать только двумерные системы. Это связано как с повышенными требованиями к вычислительным ресурсам в случае трехмерных систем, так и с простотой визуализации результатов расчетов в двумерном случае. В двумерном случае ячейка моделирования представляет собой прямоугольник. В случае периодичности в одном направлении пара противолежащих сторон отождествляется, т.е. ячейку моделирования можно представить теперь как боковую поверхность цилиндра. В случае периодичности в двух направлениях отождествляются обе пары противоположных сторон и ячейку моделирования можно теперь представить как боковую поверхность тора. Если атом выходит за пределы ячейки моделирования, то вследствие периодичности он входит в ячейку с противоположной стороны.
1.7. Начальное состояние
В данной работе будут исследоваться с помощью МД кристаллы. Рассмотрим размещение совершенного кристалла в прямоугольной ячейке моделирования в случае периодичности в одном направлении. Периодическая структура самого кристалла накладывает ограничения на размер ячейки моделирования в направлении периодичности. Действительно, если в вершине, находящейся на одной из сторон ячейки моделирования находится атом кристалла, то эквивалентный атом кристалла должен быть в эквивалентной вершине, находящейся на другой из тождественных сторон. Это приводит к ограничениям на возможную длину ячейки моделирования в направлении периодичности и возможные ориентации кристаллографических осей кристалла относительно сторон ячейки моделирования. Возможны такие ориентации кристалла, при которых указанное выше требование выполнить точно невозможно.

Рис.2 Периодичность ячеек моделирования и размещение кристалла в ячейке моделирования.
Если же ориентация кристалла выбрана удачно, то длина ячейки моделирования может принимать значения кратные некоторой величине. Однако, на самом деле, эти ограничения не очень существенны. Для всех длин ячейки моделирования и ориентаций кристалла можно найти близкие к ним значения этих величин, для которых условие будет выполняться точно. Рецепт состоит в том, чтобы просто совместить указанную эквивалентную вершину с ближайшим эквивалентным атомом кристалла.
Если есть периодичность (см. рис. 2) и по второму направлению, то должно выполняться аналогичное требование и для второго направления. При этом необходимо заметить, что ориентация второй стороны для прямоугольной ячейки моделирования уже задана, поскольку она перпендикулярна первой стороне. Поэтому её длина будет кратна некоторой величине.
Если не принять специальных мер при подготовке начального состояния системы, то в ней возникают коллективные движения - колебания. Это связано с тем, что система может оказаться в сжатом или растянутом состоянии из-за несоответствия температуры системы с постоянной кристаллической решетки. Другими словами это тепловое расширение (сжатие) системы. Такие колебания имеют большой период и слабо затухают. Накладываясь на исследуемый процесс (например, деформирование системы) они смазывают картину этого исследуемого процесса. Следовательно, от этих колебаний необходимо избавиться. Это можно сделать несколькими способами. Во-первых, подождать пока колебания затухнут. Однако из-за большого периода и малого затухания это требует большого времени. Во-вторых, попытаться подогнать постоянную решетки кристалла к температуре. Опыт показывает, что, сделав несколько попыток, можно полностью исключить колебания. В-третьих, такую подгонку можно выполнить автоматически. О том, как это можно сделать, будет сказано ниже.
В МД моделировании часто возникает необходимость иметь систему в состоянии, характеризуемом определенной температурой. Однако, как мы можем получить систему с заданной температурой? Другими словами, как мы можем контролировать систему?
Для изменения температуры необходимо так изменить скорости частиц, чтобы получить желаемую температуру. В алгоритме Верле со скоростью, обсуждаемом выше, это может быть выполнено заменой уравнения
 |
(9) |
на уравнение
 , , |
(10) |
где 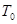 желаемая температура, и желаемая температура, и 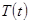 текущая температура. Такая модификация означает, что мы больше не следуем уравнениям Ньютона и, что полная энергия больше не сохраняется. текущая температура. Такая модификация означает, что мы больше не следуем уравнениям Ньютона и, что полная энергия больше не сохраняется.
1.8. Начальное состояние для кристаллов с дефектами
С помощью МД можно исследовать деформирование, как совершенных кристаллов, так и кристаллов содержащих дефекты, например, кристаллов подвергнутых облучению. О том, как подготовить начальное состояние для совершенного кристалла, было сказано выше. Подготовка начального состояния для облученного кристалла намного более сложная задача. Однако, если известны доза и спектр первично выбитых атомов, МД позволяет выполнить моделирование каскада повреждений [9,10,11]и таким образом решить эту сложную задачу. При этом описанные выше потенциалы, необходимо дополнить, чтобы учесть отталкивание на малых расстояниях, например, гладко сшивая их с потенциалом Циглера-Бирсака-Литмарка [12]. Такой подход позволяет учесть многие явления, возникающие при облучении, но является достаточно сложным и лежит за рамками данной работы.
Можно также исследовать влияние определенных дефектов, возникающие при облучении ГПУ кристаллов на их пластические свойства. Например, можно исследовать влияние межузельных кластеров и дефектов Френкеля. Очевидно, что начальные состояния, содержащие такие дефекты, легко приготовить, стартуя с начального состояния для идеального кристалла. Для этого необходимо удалить (добавить, переместить) атомы кристалла так, чтобы получилась конфигурация кристалла с требуемыми дефектами. Кристалл при этом получается обычно в напряженном состоянии. Это справедливо особенно при добавлении атомов, так как для добавленных атомов расстояния до ближайших атомов кристалла обычно намного меньше, чем равновесные расстояния между атомами в кристалле. Из-за сильного роста потенциала межатомного взаимодействия на малых расстояниях такие атомы обладают большой потенциальной энергией. Если не принять специальных мер, это может вызвать разлет кристалла. Чтобы не допустить этого и обеспечить релаксацию напряжений можно использовать процедуру минимизации и последующий подогрев системы до нужной температуры.
1.9. Нагрузка
В данной роботе рассматривалось деформирование кристаллов путем одноосного растяжения. Поскольку вдоль направления растяжения наложены периодические граничные условия, то отсутствуют свободные границы, к которым можно было бы приложить нагрузку. Поэтому задается растяжение системы, и потом находится возникшее вследствие этого напряжение. МД и деформирование выполняются одновременно. После каждого шага по времени МД выполняется малое растяжение, обеспечивающее нужную скорость деформации (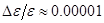 на одном шаге). Растяжение выполнялось двумя способами. В первом, традиционно используемом [13], система растягивается равномерно по длине. При этом координаты атомов вдоль направления растяжения умножаются на масштабный множитель на одном шаге). Растяжение выполнялось двумя способами. В первом, традиционно используемом [13], система растягивается равномерно по длине. При этом координаты атомов вдоль направления растяжения умножаются на масштабный множитель 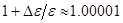 . На этот же множитель умножается длина ячейки моделирования. Согласно второму способу, предложенному в данной работе, растяжение концентрируется только возле торцов системы. Этот способ лучше соответствует экспериментальной ситуации, когда нагрузка прикладывается к торцам системы. При этом длина ячейки моделирования умножается на масштабный множитель, а координаты атомов не умножаются. . На этот же множитель умножается длина ячейки моделирования. Согласно второму способу, предложенному в данной работе, растяжение концентрируется только возле торцов системы. Этот способ лучше соответствует экспериментальной ситуации, когда нагрузка прикладывается к торцам системы. При этом длина ячейки моделирования умножается на масштабный множитель, а координаты атомов не умножаются.
1.10. Уравнение для ширины ячейки моделирования
Если боковые стороны системы по отношению к растяжению свободны, то нет необходимости следить за шириной ячейки моделирования. Если же на систему наложены периодические граничные условия по двум направлениям, то изменению ширины ячейки моделирования необходимо уделить особое внимание. При растяжении появляются сжимающие в поперечном направлении напряжения и поперечный размер (ширина системы) уменьшается. Если ширину ячейки моделирования не изменять, то появится зазор, который будет увеличиваться со временем - система разорвется в поперечном направлении.
Один из подходов [13] состоит в умножении ширины ячейки моделирования на  , при увеличении длины в , при увеличении длины в 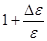 раз. Здесь раз. Здесь 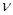 примерно равно коэффициенту Пуассона. Этого, однако, может оказаться недостаточно, поэтому, вводят дополнительную оптимизацию поперечного размера системы, основанную на методе Монте-Карло. После каждых ~20 шагов по времени МД предлагается изменение поперечного размера системы. Если в результате этого изменения энергия системы уменьшается, изменение принимается, в противном случае отклоняется. Вследствие этого, точное значение, выбранное для , становится некритичным. примерно равно коэффициенту Пуассона. Этого, однако, может оказаться недостаточно, поэтому, вводят дополнительную оптимизацию поперечного размера системы, основанную на методе Монте-Карло. После каждых ~20 шагов по времени МД предлагается изменение поперечного размера системы. Если в результате этого изменения энергия системы уменьшается, изменение принимается, в противном случае отклоняется. Вследствие этого, точное значение, выбранное для , становится некритичным.
В данной работе предложен и используется другой подход, основанный на динамическом уравнении для ширины ячейки моделирования. Выше уже было отмечено, что из-за периодичности в поперечном направлении система имеет топологию цилиндра. Сжимающие в поперечном направлении напряжения приводят к уменьшению боковой поверхности цилиндра и, следовательно, к уменьшению радиуса цилиндра. Записывая 2-ой закон Ньютона для движения системы как целого вдоль радиуса, имеем
 , , |
(11) |
где 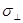 - напряжение в поперечном относительно растяжения направлении, - напряжение в поперечном относительно растяжения направлении, 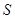 - площадь системы. Учитывая, что ширина ячейки моделирования - площадь системы. Учитывая, что ширина ячейки моделирования  , имеем для неё уравнение , имеем для неё уравнение
 , , |
(12) |
Чтобы, исключить колебательные процессы, удобно ввести в правую часть уравнения слабое фиктивное затухание 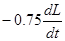 . Решая уравнения для . Решая уравнения для  на каждом временном шаге МД, мы поддерживаем ширину ячейки моделирование вблизи равновесного положения. Очевидно также, что данный подход можно использовать для демпфирования колебаний системы рассмотренных выше. на каждом временном шаге МД, мы поддерживаем ширину ячейки моделирование вблизи равновесного положения. Очевидно также, что данный подход можно использовать для демпфирования колебаний системы рассмотренных выше.
1.11. Контроль системы
Правильность работы программы МД контролировалась с помощью закона сохранения энергии:
 , , |
(13) |
где 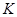 - кинетическая энергия атомов системы; - кинетическая энергия атомов системы;  - потенциальная энергия их взаимодействия; - потенциальная энергия их взаимодействия; 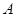 - работа, произведенная над системой. Выполнение закона сохранения энергии очень важно при исследовании пластичности твердых тел. Это связано с тем, что хотя тепловое равновесие устанавливается быстро, но установление механического равновесия требует большого времени. Поэтому при деформировании система находится в тепловом равновесии, но скорее не находится в механическом равновесии, т.е. является неравновесной. Следовательно, потеря или приход энергии, вследствие невыполнения закона сохранения энергии, может существенно повлиять на характер поведения системы при деформации. - работа, произведенная над системой. Выполнение закона сохранения энергии очень важно при исследовании пластичности твердых тел. Это связано с тем, что хотя тепловое равновесие устанавливается быстро, но установление механического равновесия требует большого времени. Поэтому при деформировании система находится в тепловом равновесии, но скорее не находится в механическом равновесии, т.е. является неравновесной. Следовательно, потеря или приход энергии, вследствие невыполнения закона сохранения энергии, может существенно повлиять на характер поведения системы при деформации.
Неточное сохранение энергии связано в основном с ошибками, возникающими из-за конечного шага интегрирования по времени, а также с ошибками, возникающими из-за конечной точности представления чисел в компьютере.
Оба типа ошибок можно уменьшить, уменьшая шаг интегрирования по времени, что, однако, увеличивает время вычислений.
Другой тип ошибок возникает из-за использования потенциала с обрезанием. Скачок потенциала на радиусе обрезания при пластической деформации, когда атомы могут двигаться друг относительно друга, приводит к значительному нарушению закона сохранении энергии. Использование потенциала без скачка (3) позволяет существенно улучшить выполнения закона сохранения энергии. Потенциал (3), однако, имеет скачок производной (силы) на радиусе обрезания 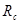 . Это также приводит к несоблюдению закона сохранения энергии. Оно особенно ярко проявляется при уменьшении радиуса обрезания от канонических значений . Это также приводит к несоблюдению закона сохранения энергии. Оно особенно ярко проявляется при уменьшении радиуса обрезания от канонических значений 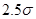 и и 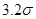 . Это связанно с тем, что канонические значения радиуса обрезания находятся в минимумах радиального распределения атомов гексагональной решетки. Когда же попадает в максимум радиального распределения число атомов, то испытывающих действие силы (при . Это связанно с тем, что канонические значения радиуса обрезания находятся в минимумах радиального распределения атомов гексагональной решетки. Когда же попадает в максимум радиального распределения число атомов, то испытывающих действие силы (при 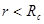 ), то прекращающих испытывать ее действие (при ), то прекращающих испытывать ее действие (при 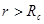 ), становиться очень большим, что и приводит к существенному несохранению энергии. Чтобы избавиться от скачка производной потенциала на радиусе обрезания потенциал был модернизирован. Пусть ), становиться очень большим, что и приводит к существенному несохранению энергии. Чтобы избавиться от скачка производной потенциала на радиусе обрезания потенциал был модернизирован. Пусть
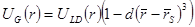 , , |
(14) |
где
 |
(15) |
и 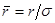 , , 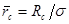 , , 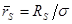 . Тогда модернизированный потенциал имеет вид . Тогда модернизированный потенциал имеет вид
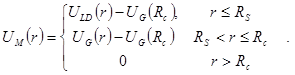 |
(16) |
Модернизированный потенциал гладко сшивается (до второй производной) с потенциалом Леннарда-Джонса на радиусе сшивки  и зануляетсявместе со своей первой производной на радиусе обрезания . С этим потенциалом при значениях параметров и зануляетсявместе со своей первой производной на радиусе обрезания . С этим потенциалом при значениях параметров 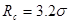 и и 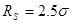 были проведены все расчеты в данной работе. были проведены все расчеты в данной работе.
1.12. Вычисление физических величин
При деформировании системы все физические величины, такие как напряжение 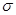 ,температура ,температура 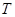 , кинетическая энергия , кинетическая энергия 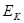 , потенциальная энергия , потенциальная энергия  характеризующие деформируемую систему меняются. Их мгновенные значения, усредненные по малым промежуткам времени чтобы исключить тепловые колебания, описывают состояние деформируемой системы. В отличие от равновесных систем мы не можем теперь использовать усреднение по времени, а должны использовать усреднение по различным начальным состояниям системы. характеризующие деформируемую систему меняются. Их мгновенные значения, усредненные по малым промежуткам времени чтобы исключить тепловые колебания, описывают состояние деформируемой системы. В отличие от равновесных систем мы не можем теперь использовать усреднение по времени, а должны использовать усреднение по различным начальным состояниям системы.
Кинетическая и потенциальная энергия находятся как
 |
(17) |
 |
(18) |
Температура определяется как
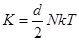 , , |
(19) |
где  - размерность системы. В двухмерном случае - размерность системы. В двухмерном случае  - средней кинетической энергией. Выражение для тензора напряжений, основанное на вириальной теореме [14,15], имеет вид - средней кинетической энергией. Выражение для тензора напряжений, основанное на вириальной теореме [14,15], имеет вид
 , , |
(20) |
где 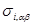 - - 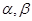 -компоненты тензора напряжений для атома -компоненты тензора напряжений для атома 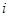 , , 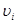 - объем, приходящийся на атом ( - объем, приходящийся на атом (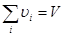 , где , где 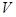 - полный объем системы), - полный объем системы), 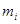 - масса атома , - масса атома , 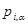 - -  -компонента его импульса, -компонента его импульса, 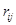 - расстояние между атомами и - расстояние между атомами и 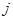 ( (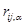 - компонента вектора, направленного от -го атома к -му атому). Это выражение для тензора напряжений не единственное, существуют и другие его определения. Однако, когда напряжения усредняются по объему различные определения быстро сходятся к макроскопическому полю напряжений. Во время моделирования кривые напряжение - деформация строятся после усреднения атомного напряжения по всей системе. - компонента вектора, направленного от -го атома к -му атому). Это выражение для тензора напряжений не единственное, существуют и другие его определения. Однако, когда напряжения усредняются по объему различные определения быстро сходятся к макроскопическому полю напряжений. Во время моделирования кривые напряжение - деформация строятся после усреднения атомного напряжения по всей системе.
1.13. Визуализация
МД позволяет получать огромные объемы информации, описывающие исследуемую систему во всех деталях. Поэтому возникает задача, извлечь из этого моря информации нужную информацию и предоставить ее в виде, удобном для восприятия. Например, увидеть дефекты находящиеся внутри трехмерного кристалла, невозможно, поскольку их закрывают атомы, находящиеся на том же луче зрения, но ближе к наблюдателю. Однако, оставив только атомы, окружающие дефекты и удалив все остальные, это можно легко сделать. Другая возможность состоит в использовании анимации для исследования временной эволюции деформируемой системы.
Во время деформирования кристаллов упорядоченное расположение атомов кристалла нарушается, появляются дефекты. Для исследования локального атомного порядка обычно используется алгоритм, известный как Common Neighbor Analysis (CNA) [16,17]. Для того чтобы определить структуру кристалла в этом алгоритме исследуются связи между атомами и его соседями. Два атома считаются связанными, если расстояние между ними меньше критического расстояния, выбранного между первыми двумя пиками в радиальной функции распределения. Связи классифицируются с помощью трех целых чисел  . Первое из них, , есть число общих соседей, т.е. атомов, связанных с обоими атомами в рассматриваемой связи. Второе, , есть число связей между этими общими соседями. Третье, . Первое из них, , есть число общих соседей, т.е. атомов, связанных с обоими атомами в рассматриваемой связи. Второе, , есть число связей между этими общими соседями. Третье, 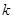 , есть самая длинная цепочка, которую можно образовать из этих связей. , есть самая длинная цепочка, которую можно образовать из этих связей.
Число и тип связей, которые имеет атом, определяют локальную кристаллическую структуру. Например, атомы в совершенном ГЦК кристалле имеет 12 связей типа 421, тогда как в совершенном ГПУ кристалле имеют 6 связей типа 421 и 6 типа 422.
Использование CNA позволяет сделать видимыми при моделировании дислокации, границы зерен и дефекты упаковки. Например, при деформировании кристаллов меди, с помощью CNA классифицируют атомы на три класса: ГЦК, ГПУ и “другие”, где в класс “другие” попадают атомы, которые имеют число связей, отличное от 12, или тип их связей отличен от 421 и 422. Тогда внутренние дефекты упаковки видны как две сопряженные (111) плоскости ГПУ атомов, несвойственные дефекты упаковки видны как две (111) плоскости атомов ГПУ, разделенных (111) плоскостью атомов ГЦК. Границы двойников видны как одиночные (111) плоскости ГПУ атомов. Ядра дислокации и границы зерен состоят из атомов класса “другие”, хотя границы зерен содержат также немного ГПУ атомов.
Тепловые колебания атомов мешают выполнять CNA. Если при моделировании температура сравнительно низкая, достаточно тщательно выбрать критическое расстояние. Для высоких же температур необходимо предварять CNA короткой минимизацией (достаточной чтобы уменьшить тепловые колебания атомов, но избежать движения дислокаций).
В двухмерных системах нет нужды выполнять CNA анализ - дефекты видны непосредственно. Кроме того, в отличие от трехмерных систем, отсутствуют заслоняющие атомы. Поэтому двухмерные системы удобны для анимации, т.е. воспроизведения временной эволюции деформируемой системы. Для создания анимации через заданное число шагов по времени МД, используя координаты атомов, формировалося изображение системы, которое затем сохранялось на жестком диске в bmp-файле. Анимация достигалась выводом этих изображений на экран дисплея в той же последовательности, в которой они создавались. Одно из преимуществ анимации - это наглядность. Поля других физических величин, например, напряжения, температуры, используя подходящую кодировку, также можно использовать для анимации.
2. Моделирования пластической деформации ГПУ-кристаллов
Автором была создана программа для изучения пластичности в двумерных кристаллах. Двумерные системы были выбраны, чтобы обойти проблемы, связанные с высокими требованиями к вычислительным ресурсам в случае трехмерных систем и визуализацией результатов вычислений. Для решения уравнений движения использовался алгоритм Верле со скоростью. В качестве потенциала взаимодействия между атомами был выбран потенциал Леннарда-Джонса. При моделировании вычислительная ячейка растягивалась вдоль умножением ее продольного размера на каждом шаге по времени на масштабный множитель (1+ε), где ε – малое число (0.00001), выбранное так, чтобы обеспечить требуемую скорость деформации. Координаты атомов при этом не менялись, т.е. при этом вводился зазор между атомами в смежных ячейках моделирования. При этом нагрузка прикладывалась к торцам кристалла, что лучше соответствует эксперименту. Поперечный размер системы контролировался с помощью динамического уравнения (12). Перед деформацией система приводилась в равновесное состояние с заданной температурой коротким прогоном с помощью МД. При вычислении кривых напряжение-деформация проводилось усреднение напряжения по атомам всей системы. С помощью закона сохранения энергии контролировалась правильность работы программы. Для наблюдения за временной эволюцией деформируемого кристалла использовалась анимация. Блок-схема программы приведена ниже.
Задание входных данных
· Шаг по времени
· Число шагов по времени
· Число атомов по x (направление деформации)
· Число атомов по y
· Скорость деформации ε
· Температура
· Тип решетки (гексагональная)
· Ориентация решетки
· Граничные условия (периодические)
· Потенциал
|
Задание начальных значений
· Начальные смещения атомов
· Начальные скорости атомов
|
| Достижение равновесного состояния с заданной температурой |
| Цикл по атомам (Вычисление начального ускорения) |
Сила Fi
, действующая на i-тый атом = сумме сил, действующих со стороны соседних атомов.
Ускорение i-того атома ai
(t) = Fi
/mi
|
| Конец цикла по атомам |
| Цикл по времени (Траектория + растяжение) |
| Цикл по атомам (Вычисление нового положения и промежуточной скорости) |
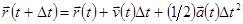
|
| Конец цикла по атомам |
| Проверка граничных условий |
| Растяжение |
| Цикл по атомам (Вычисление нового ускорения и новой скорости ) |
Сила Fi
, действующая на i-тый атом = сумме сил, действующих со стороны соседних атомов.
Ускорение i-того атома ai
(t+Δt) = Fi
/mi
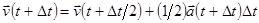
|
| Конец цикла по атомам |
Контроль параметров
· деформация
· напряжение
· температура
|
| Конец цикла по времени |
| Визуализация |
Программа позволяет проводить как обычную МД, так и использовать процедуру минимизации. Программа позволяет исследовать влияние ориентации кристалла, скорости деформирования и температуры на процесс деформирования. Она также позволяет использовать свободные и периодические граничные условия на боковых, относительно растяжения, сторонах кристалла.
С помощью программы проведено моделирование пластического деформирования двумерных кристаллов с гексагональной решеткой. Такие решетки характерны для базисной плоскости ГПУ кристаллов и для плоскости (111) ГЦК кристаллов. Начальная температура  0.025 0.025 . На рис.3 приведена сумма полной энергия кристалла и произведенной над ним работы (на один атом кристалла) как функция времени. Видно, что с точностью до 5 знака эта величина сохраняется, что свидетельствует о правильности работы программы. . На рис.3 приведена сумма полной энергия кристалла и произведенной над ним работы (на один атом кристалла) как функция времени. Видно, что с точностью до 5 знака эта величина сохраняется, что свидетельствует о правильности работы программы.

Рис.3 Зависимость сумма полной энергия кристалла и произведенной над ним работы (на один атом кристалла) как функция времени.
На рис.4 приведено напряжение, поперечное относительно направления растяжения, как функция времени. Видно, что оно почти все время поддерживается невысоким. Это свидетельствует о правильности выбранного алгоритма регулирования ширины ячейки моделирования.

Рис.4 Зависимость напряжения, поперечного относительно направления растяжения, от времени.
Наконец, на рис.5 приведена кривая напряжение-деформация. Эта кривая полностью соответствует экспериментальным кривым напряжение-деформация для совершенных кристаллов.
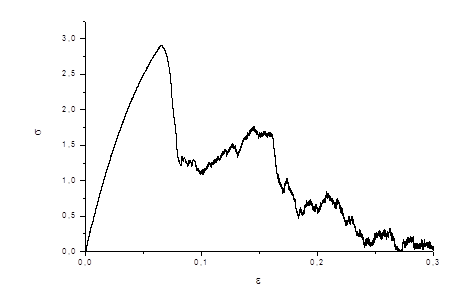
Рис.5 Кривая растяжения совершенного кристалла
На этой кривой можно выделить несколько стадий в деформировании кристалла [18,19]. На первой, упругой стадии, происходит накопление энергии. Затем, по мере роста напряжения кристалл переходит в нелинейную область, в конце которой появляются дислокации. Начинается сброс напряжения, который заканчивается появлением полос скольжения, которые и обеспечивают дальнейший сброс напряжения. Затем происходит частичное упрочнение кристалла с последующими сбросами напряжения. На этой стадии происходит накопление вакансий, их слияние в вакансионные поры. На последней стадии поры, объединяясь, дают начало трещине, которая, развиваясь, разрывает кристалл. На рис.6-11 даны изображения кристалла, которые иллюстрируют описанную выше стадийность пластического деформирования и разрушения кристалла.
Заключение
1. Изучен метод молекулярной динамики со всеми его важнейшими ингредиентами: потенциал взаимодействия, граничные условия, алгоритм интегрирования по времени, задание начальных условий, контроль термодинамических параметров в процессе моделирования, контроль достижения термодинамического равновесия, измерение физических величин. Изучены особенности применения метода молекулярной динамики к исследованию пластичности реакторных материалов.
2. Создана программа для изучения пластичности в кристаллах. Создана программа визуализации процесса пластической деформации и разрушения кристаллов. Предложен новый подход к моделированию растяжения кристаллов, близкий к используемому в эксперименте. Предложено динамическое уравнение для поперечного размера ячейки моделирования.
3. Проведено моделирование развития пластической деформации ГПУ кристаллов при одноосном растяжении. Показана принципиальная возможность имитации с помощью этого метода кривых растяжения совершенных кристаллов, изменения температуры образца, появления дислокаций, полос скольжения, одиночных вакансий и их скоплений, а также процесса разрушения кристаллов.
Список использованных источников
1. В.В.Кирсанов, ЭВМ-эксперимент в атомном материаловедении, Энергоатомиздат, 1990.
2. J.Schiotz, Scripta Mater., 51, 837 (2004).
3. K.W.Jacobsen, J.K.Norskov, M.J.Puska, Phys. Rev. B35, 7423 (1987).
4. K.W.Jacobsen, P.Stoltze, J.K.Norskov, Surf. Sci. 366, 394 (1996).
5. L.Verlet, Phys. Rev. 159, 98 (1967); Phys. Rev. 165, 201 (1967).
6. D. van der Spoel, E. Lindahl, B. Hess at al. Gromacs User Manual, v. 3.2, University of Groningen.
7. K. Refson. Moldy User’s Manual, v. 2.16, (2001).
8. W.Smith, M.Leslie, T.R.Forester. The DL_POLY_2 User Manual, (2003), Daresbery Laboratory.
9. J.B.Gibson, A.N.Goland, M.Milgram, G.H.Vineyard, Phys. Rev. 120, 1229 (1960).
10. M.P.Allen, D.J.Tildesley Computer simulation of liquids. Clarendon Press, Oxford, 1989.
11. K.Nordlund, Comput. Mater. Sci., 3, 448 (1995).
12. J.F.Ziegler, J.P.Biersack, U.Littmark, The stopping and range of ions in solids. Pergamon Press, N.Y., 1987.
13. J.Schiotz, T.Vegge, F. D. Di Tolla, K.W.Jacobsen, Phys. Rev. B60, 11971 (1999),cond-mat/9902165.
14. T.Egami, K.Maede, and V.Vitek, Phil. Mag. A 41, 883 (1980).
15. J.R.Ray and A.Rahman, J. Chem. Phys. 80, 4423 (1984).
16. H.Jonsson and H.C.Andersen, Phys. Rev. Lett. 60, 2295 (1988).
17. A.S. Clarke and H.Jonsson, Phys. Rev. E 47, 3975 (1993).
18. Р.Хоникомб Р. Пластическая деформация металлов. М. Мир. 1972.
19. И.М.Неклюдов, Н.В.Камышанченко. Физические основы прочности и пластичности металлов. ч. 3. Пластическая деформация и разрушение кристаллических материалов. Изд. “Педагогика-Пресс” и Белгородского государственного университета, Белгород, 2001
|