| Прогнозирование энтропий органических соединений методом статистической термодинамики [1, 13]
Применение квантовой теории к описанию энергетических соотношений молекул газов привело к развитию достаточно надежных методов расчета термодинамических свойств веществ, находящихся в состоянии идеального газа. Для энтропии это методы статистической термодинамики [13], где свойство представляется в виде суммы вкладов для различных видов движения молекул.
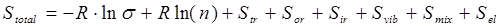 . (2.12) . (2.12)
Здесь необходимо остановиться более подробно на методике расчета отдельных вкладов.
 - здесь это вклад в энтропию, обусловленный симметрией наружного вращения молекулы как целого и симметрией внутреннего вращения только тех групп, для которых она не может быть учтена при анализе энтропийного вклада, обусловленного внутренним вращением. Детали расчета симметрии наружного вращения молекул рассмотрены выше. Второй вклад, о котором идет здесь речь, касается внутреннего вращения в молекуле, которое обеспечивается не одной, а одновременно двумя связями. Например, для ароматических систем такими группами являются п-фениленовые фрагменты (–С6Н4–), внутреннее вращение которых происходит одновременно по двум связям. - здесь это вклад в энтропию, обусловленный симметрией наружного вращения молекулы как целого и симметрией внутреннего вращения только тех групп, для которых она не может быть учтена при анализе энтропийного вклада, обусловленного внутренним вращением. Детали расчета симметрии наружного вращения молекул рассмотрены выше. Второй вклад, о котором идет здесь речь, касается внутреннего вращения в молекуле, которое обеспечивается не одной, а одновременно двумя связями. Например, для ароматических систем такими группами являются п-фениленовые фрагменты (–С6Н4–), внутреннее вращение которых происходит одновременно по двум связям.
Учет вклада на смешение вращательных изомеров ( ), обусловленное наличием в молекуле хиральных центров, производится в соответствии с подходом, изложенным в п. 2.1. ), обусловленное наличием в молекуле хиральных центров, производится в соответствии с подходом, изложенным в п. 2.1.
Вклад поступательного движения молекул в энтропию 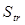 рассчитывается по уравнению рассчитывается по уравнению
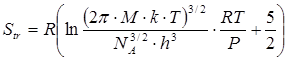 , (2.13) , (2.13)
где М – молекулярная масса;  - мольный объем газа; - мольный объем газа;  - число Авогадро; h – постоянная Планка. Подставив в это уравнение значения констант k, h, NA
, при стандартном состоянии P = 1 атм = 101325 Па, получим формулу, удобную для практических расчетов энтропии поступательного движения: - число Авогадро; h – постоянная Планка. Подставив в это уравнение значения констант k, h, NA
, при стандартном состоянии P = 1 атм = 101325 Па, получим формулу, удобную для практических расчетов энтропии поступательного движения:
 Дж/(моль·К), (2.14) Дж/(моль·К), (2.14)
где Р должно выражаться в физических атмосферах, а остальные величины – в соответствии с системой СИ.
Таким образом, для вычисления вкладов поступательного движения молекул в энтропию газа нужно знать только его молекулярную массу, и для изомеров эти вклады равны по определению.
Остальные вклады требуют привлечения информации о геометрии, энергетических характеристиках молекул и частотах колебательного спектра. Для получения подобной информации используются различные расчетные методы. Окончательная обработка информации и вычисление энтропийных вкладов выполняются с помощью разработанной на кафедре ТО и НХС СамГТУ программы Entropy, описание которой приведено далее.
В рамках программы Entropy геометрия молекулы оптимизируется методом молекулярной механики (силовое поле MMX на базе силового поля Эллинджера MM2) программой PCModel 3.2. Для оптимизации молекул бифенилов желательно использовать PCModel 4.0, обладающую большими возможностями при расчетах в p-электронных системах. Выходной информацией являются оптимизированная геометрия молекулы для наиболее устойчивого конформера и информация об изменении энергии молекулы при вращении каждого из волчков, сохраняемые в отдельных файлах. Для формирования потенциальной кривой барьера вращения каждого из волчков используются значения потенциальной энергии молекулы при изменении двугранного угла между избранными связями волчка и остова от 0о
до 360о
с шагом 10о
. При этом на каждом фиксированном значении угла проводится оптимизация геометрии молекулы.
На основании сведений о геометрии молекулы рассчитывается произведение главных центральных моментов инерции IA
IB
IC
, являющееся свободным членом кубического уравнения
 , ,
где 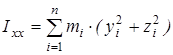 ; ; 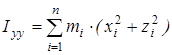 ; ;  ; ;  ; ; 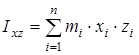 ; ; 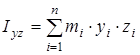 - моменты инерции молекулы (здесь n – число атомов в молекуле; mi
– масса i-того атома; xi
, yi
, zi
– координаты i-того атома в системе координат с центром, находящемся в центре инерции молекулы). Отсюда - моменты инерции молекулы (здесь n – число атомов в молекуле; mi
– масса i-того атома; xi
, yi
, zi
– координаты i-того атома в системе координат с центром, находящемся в центре инерции молекулы). Отсюда
 . (2.15) . (2.15)
В дальнейшем рассчитывается сумма состояний жесткого ротатора
 (2.16) (2.16)
и вклад в энтропию, обусловленный вращением молекулы как целого
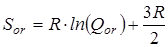 , (2.17) , (2.17)
где h – постоянная Планка, k – постоянная Больцмана.
В тех случаях, когда это предусмотрено решаемой задачей, рассчитывается вклад в энтропию, обусловленный смешением конформеров. Программой Entropy наряду с классическим подходом предусмотрен следующий вариант расчета энтропии смешения конформеров. На основании полученных ранее сведений об изменении энергии молекулы при вращении каждой из ее групп вычисляется
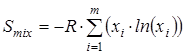 , (2.18) , (2.18)
где m – общее количество рассматриваемых конформаций (в нашем случае учитывались все состояния, полученные при повороте волчка от 0о
до 350о
с шагом 10о
, то есть m=36∙n, где n – число вращающихся групп в молекуле), xi
– мольная доля каждой конформации
 , (2.19) , (2.19)
где n – число вращающихся групп в молекуле, m – количество рассматриваемых конформаций, Ei
– энергия молекулы, в данном состоянии равная 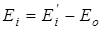 , где , где  - исходное значение энергии, - исходное значение энергии,  - наименьшая энергия молекулы, полученная при вращении всех возможных волчков. - наименьшая энергия молекулы, полученная при вращении всех возможных волчков.
Для нахождения вклада в энтропию, обусловленного колебательным движением, используются расчетные значения частот колебательного спектра, рассчитанные любым из квантово-химических методов, реализованных в программах Gaussian или Hyperchem, для оптимизированной тем же методом геометрии молекулы. Критерием качества оптимизации служит отсутствие в спектре отрицательных значений частот.
Расчет вклада в энтропию, обусловленного колебательным движением, производится следующим образом.
 , (2.20) , (2.20)
где νi
– частота из принятого к расчету набора, m – количество частот в наборе. Из полного набора частот колебательного спектра исключаются крутильные колебания, соответствующие вращению групп, участвующих в расчете вклада в энтропию от заторможенного вращения; таким образом, 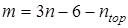 , где n – число атомов в молекуле, ntop
– число волчков. При отсутствии надежных методик определения крутильных колебаний в спектре применяется приближенная оценка типов колебаний с использованием режима Animate программы HyperChem 5.0. , где n – число атомов в молекуле, ntop
– число волчков. При отсутствии надежных методик определения крутильных колебаний в спектре применяется приближенная оценка типов колебаний с использованием режима Animate программы HyperChem 5.0.
Информация о геометрии молекулы и потенциальных кривых барьеров вращения волчков используется для расчета вклада в энтропию, обусловленного внутренним вращением групп в молекуле. Энтропийный вклад определялся как
 , (2.21) , (2.21)
здесь n – число максимумов потенциальной кривой барьера вращения группы, s– число симметрии группы (подходы к определению чисел симметрии вращающихся групп рассмотрены выше), Sfr
– энтропия свободного вращения волчка, 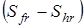 - разность между энтропиями свободного и заторможенного вращения, определяемая по таблицам Питцера и Гуинна [1] как функция - разность между энтропиями свободного и заторможенного вращения, определяемая по таблицам Питцера и Гуинна [1] как функция  и и  , где Vo
– эффективный барьер вращения волчка, Qfr
– статистическая сумма по состояниям свободного внутреннего вращения. , где Vo
– эффективный барьер вращения волчка, Qfr
– статистическая сумма по состояниям свободного внутреннего вращения.
Величина эффективного барьера вращения принимается равной 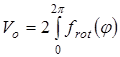 , где , где  - зависимость изменения потенциальной энергии молекулы от угла поворота волчка φ. Для расчета Vo
полученные методом молекулярной механики значения потенциальной энергии молекулы при заданных значениях угла поворота волчка описываются с помощью кубического сплайна, затем полученный сплайн интегрируется по методу Симпсона. - зависимость изменения потенциальной энергии молекулы от угла поворота волчка φ. Для расчета Vo
полученные методом молекулярной механики значения потенциальной энергии молекулы при заданных значениях угла поворота волчка описываются с помощью кубического сплайна, затем полученный сплайн интегрируется по методу Симпсона.
Статистическая сумма по состояниям свободного внутреннего вращения рассчитывалась как
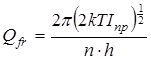 , (2.22) , (2.22)
где Iпр
– приведенный момент инерции волчка, который рассчитывался в соответствии со следующей процедурой.
Для вращающейся группы вводится координатная система с осями x, y, z, расположенными следующим образом: ось z совпадает с осью вращения волчка, ось x проходит через центр масс волчка и перпендикулярна оси z, ось y проходит через точку пересечения осей x, z и перпендикулярна к ним. Атомы волчка, лежащие на оси z, из дальнейшего рассмотрения исключаются. Далее производится расчет следующих величин:  - момент инерции волчка относительно оси z, - момент инерции волчка относительно оси z,  и и  - произведения моментов инерции, - произведения моментов инерции, 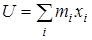 - фактор несбалансированности волчка. - фактор несбалансированности волчка.
Затем находятся направляющие косинусы осей x, y, z относительно главных центральных осей 1, 2, 3 инерции молекулы. Направление осей выбирается таким образом, чтобы обе системы координат были или правыми, или левыми. При этом должно соблюдаться условие равенства единице определителя матрицы направляющих косинусов, т.е.
 , ,
что может использоваться для проверки правильности определения направляющих косинусов.
Приведенный момент инерции рассчитывается следующим образом:
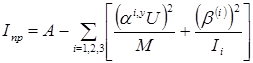 , (2.23) , (2.23)
где 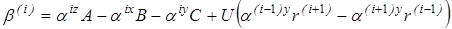 . Здесь r(
i)
– проекции на главные оси инерции молекулы вектора, направленного из центра тяжести молекулы в центр координат волчка, индекс i принимает значения 1, 2, 3 в циклическом порядке, т.е. при i=1 индекс i-1 равен 3, а индекс i+1 при i=3 равен 1. . Здесь r(
i)
– проекции на главные оси инерции молекулы вектора, направленного из центра тяжести молекулы в центр координат волчка, индекс i принимает значения 1, 2, 3 в циклическом порядке, т.е. при i=1 индекс i-1 равен 3, а индекс i+1 при i=3 равен 1.
Достоверность полученных значений энтропии определяется надежностью расчетных процедур. Значения энтропийных вкладов зависят от набора методов, с помощью которых они вычисляются. Для простых органических молекул с достаточной долей вероятности можно признать достоверными все описанные выше процедуры расчета вкладов. Для более сложных соединений могут возникать неоднозначные ситуации при определении большинства вкладов в энтропию, особенно энтропии смешения конформеров, и вопросы, связанные с принципиальной возможностью заторможенного вращения объемных волчков. В этом случае принятию решений должен предшествовать обстоятельный анализ экспериментальных сведений по энтропиям родственных соединений.
При наличии в молекуле соединения объемных волчков, вращение которых практически невозможно, вклад на внутреннее вращение таких волчков исключается из расчета. В этом случае при расчете вклада  (уравн. 2.20) из полного набора частот колебательного спектра не исключаются крутильные колебания, соответствующие вращению рассматриваемых групп. Очевидно, что для принятия подобных решений необходим достаточный опыт. (уравн. 2.20) из полного набора частот колебательного спектра не исключаются крутильные колебания, соответствующие вращению рассматриваемых групп. Очевидно, что для принятия подобных решений необходим достаточный опыт.
2.5. Описание приложения
Entropy
С целью максимального упрощения приведенной процедуры расчета вкладов в энтропию было создано приложение Entropy. Приложение разработано в среде Delphi 5 и предназначено для работы в операционных системах Windows (начиная с версии Windows 95) и Windows NT (начиная с версии 4.0). Работа приложения в указанных операционных системах протекает стабильно.
Расчет составляющих энтропии производится в полном соответствии с расчетными процедурами, изложенными в разделе 2.4. Программная реализация значительной части приведенных алгоритмов была произведена к.х.н. А.А. Пимерзиным в среде Quick Basic 4.5 при разработке MS-DOS приложения для расчета энтропийных вкладов. И.А. Нестеровым была выполнена [25] адаптация процедур к языку Object Pascal с проведением оптимизации всех алгоритмов для наилучшего их функционирования на данном наборе задач и в разработанной автором структуре приложения.
Исходными данными для работы приложения являются:
- файл, содержащий сведения о геометрии изучаемой молекулы, в формате MMX (создается PCModel);
- файлы, содержащие сведения о потенциальных кривых барьеров вращения волчков молекулы, в формате XYZ PCModel версии 3.0 (создается PCModel или конвертируется из ее log-файла);
- файл, содержащий набор частот колебательного спектра (log-файл HyperChem, полученный при выполнении команды Vibrations, или выходной файл Gaussian, содержащий информацию о частотах ИК-спектра).
В начале работы с приложением пользователь производит загрузку файла, содержащего сведения о геометрии рассматриваемой молекулы, при этом осуществляется отображение структуры молекулы в окне приложения и расчет произведения главных моментов инерции данной структуры (рис. 2.3).
Пользователь по своему желанию для большего удобства работы со структурой может выполнить ее поворот в пространстве, масштабирование или перемещение в плоскости. Все эти операции производятся перемещением мыши при нажатой левой кнопке после выбора соответствующего пункта из меню Изображение. Также пользователем может быть избран один из вариантов отображения подписей атомов (обозначения атомов, номера атомов или их ММХ-типы) при выборе одного из пунктов меню Обозначения.
Выбор волчков, принимающих участие в расчете, производится нажатием правой кнопки мыши при нахождении курсора над связью, по которой осуществляется вращение волчка. При этом программно осуществляется проверка возможности вращения по данной связи, связь должна быть одинарной и не входить в цикл.
Проверка вхождения связи в цикл осуществляется следующим образом: пусть молекула представлена в виде графа и А1
и А2
- его вершины, соответствующие атомам, образующим данную связь; исключается ребро А1
- А2
; далее производится обход графа при использовании в качестве начальных вершин последовательно А1
и А2
; совпадение наборов вершин, полученных при обходе графа как из А1
, так и из А2
, свидетельствует о вхождении связи в цикл.

Рис. 2.3. Отображение структуры молекулы 2-третбутилфенола

Рис. 2.4. Определение вращающейся группы
Если вращение по связи признается возможным и с каждым из атомов, образующих связь, связан, по крайней мере, еще один атом, то осуществляется автоматическое добавление к списку волчков избранной структуры наименее массивного из прилегающих к связи фрагментов. При этом производится расчет приведенного момента инерции волчка. Пользователем выполняется ввод названия волчка (рис. 2.4).
После ввода названия осуществляется переход к форме ввода параметров волчка, в которой следует произвести открытие файла с энергиями потенциальной кривой барьера вращения. При этом производится вывод изображения кривой, подсчет числа максимумов и расчет значения приведенного барьера вращения. Дополнительно к значению барьера, рассчитанному в соответствии с рекомендациями раздела 4.2, выводится значение 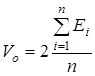 (рис. 2.5). (рис. 2.5).

Рис. 2.5. Форма для ввода параметров вращающейся группы

Рис. 2.6. Просмотр значений на потенциальной кривой вращения группы
В форме параметров волчка пользователь может изменить значения числа симметрии и числа максимумов волчка, величину барьера вращения, внеся значения в соответствующие поля формы. Масштабирование потенциальной кривой осуществляется вводом масштабного коэффициента и нажатием кнопки Применить. Также в данной форме возможен просмотр потенциальной кривой в энергетических координатах исходного XYZ-файла, для чего следует сбросить флажок Приведение к нулю.
В обеих системах координат доступен просмотр каждого из значений энергии на потенциальной кривой (рис. 2.6). Текущий вид потенциальной кривой может быть сохранен в файле формата BMP или WMF.
Форма параметров волчка доступна для внесения необходимых изменений после первого ввода информации путем выбора имени волчка из меню Волчки. После ввода параметров всех вращающихся групп возможен ввод информации о частотах колебательного спектра, который осуществляется выбором из меню Счет пункта Набор частот.
 
Рис. 2.7. Форма ввода частот Рис. 2.8. Форма ввода набора
колебательного спектра температур
В форме ввода набора частот пользователь должен открыть log-файл HyperChem с записью колебательного спектра, после чего в таблице будут выведены номера частот и их значения в см-1
. Выбором номера частоты и нажатием кнопки Удалить производится исключение частоты из спектра (рис. 2.7).
На этом завершается ввод необходимых исходных данных. Для проведения расчета следует только с помощью пункта Набор температур меню Счет задать значения температур, для которых должен быть проведен расчет составляющих энтропии. Форма ввода температур, представленная на рис. 2.8, не требует дополнительных пояснений. С ее помощью в набор температур могут быть введены как индивидуальные значения, так и наборы значений с постоянным шагом.
После ввода набора температур выбором пункта Расчет энтропийных вкладов меню Счет выполняется окончательный расчет составляющих энтропии. При этом вывод результатов производится как в форме суммарных вкладов для вещества, так и, для вкладов, обусловленных внутренним вращением и смешением конформеров, с расчетом индивидуального вклада в энтропию от каждого из волчков (рис. 2.9, 2.10). Информация, представленная в таблицах, может быть как полностью, так и частично скопирована для дальнейшего использования при проведении расчетов с помощью приложений, способных осуществлять получение данных из буфера обмена.

Рис. 2.9. Вывод итоговой информации

Рис. 2.10. Вывод информации о расчете составляющих энтропии об индивидуальных вкладах волчков в составляющие энтропии
Пример 2.6
Методом статистической термодинамики рассчитать  2,2,4-триметилпентана при температурах 298, 300, 400, 500 и 600 К. 2,2,4-триметилпентана при температурах 298, 300, 400, 500 и 600 К.
Решение
1. Структурная формула молекулы 2,2,4-триметилпентана:

2. Вклад на симметрию 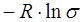 молекулы рассчитывается исходя из симметрии наружного вращения 2,2,4-триметилпентана: –R×ln(1). молекулы рассчитывается исходя из симметрии наружного вращения 2,2,4-триметилпентана: –R×ln(1).
3. В программе PCModel 3.0 создается, оптимизируется и записывается в формате MMX файл, содержащий сведения о геометрии изучаемой молекулы – 224-tmp.mmx.
4. Там же (PCModel 3.0) создаются файлы в формате XYZ, содержащие сведения о потенциальных кривых барьеров вращения волчков в молекуле:
трет-бутильный волчок – t-Bu.xyz; изопропильный волчок – i-Pr.xyz;
метильные волчки в трет-бутильном заместителе – Me1(tbu).xyz, Me2(tbu).xyz, Me3(xyz); метильные волчки в изопропильном заместителе – Me4(ipr).xyz, Me5(ipr).xyz.
5. В программе HyperChem создается файл, полученный при выполнении команды Vibrations и содержащий набор частот колебательного спектра 224-tmp.log.
6. В приложении Entropy с использованием подготовленных ранее файлов и в соответствии с рекомендациями разд. 2.5. рассчитываются составляющие энтропии. Необходимые для расчета молекулярные данные для 2,2,4-триметилпентана приведены в табл. 2.11. В ней даны: произведение главных центральных моментов инерции (IA
IB
IC
) для наиболее устойчивого конформера; потенциальные барьеры вращения (Vr
) и приведенные моменты инерции волчков (Ir
); количество максимумов на потенциальных кривых вращения (nmax
) и числа симметрии волчков (σ); номера частот, отвечающих за крутильные колебания волчков, исключенные из расчета колебательного вклада.
Таблица 2.11
Молекулярные данные 2,2,4-триметилпентана
| Волчок
|
IAIBIC·10112 ,
г3/см6
|
Vr, Дж/моль
|
Ir·1040, г/см2
|
nmax
|
σ
|
ν
|
| t-Bu
|
1,575418
|
12472,08
|
80,51
|
6
|
3
|
1
|
| i-Pr
|
21483,39
|
69,39
|
6
|
1
|
2
|
| Me1(t-Bu)
|
15523,51
|
5,324
|
3
|
3
|
4
|
| Me2(t-Bu)
|
12952,77
|
5,352
|
3
|
3
|
6
|
| Me3(t-Bu)
|
12859,60
|
5,334
|
3
|
3
|
7
|
| Me4(i-Pr)
|
12108,83
|
5,331
|
5
|
3
|
3
|
| Me5(i-Pr)
|
15357,90
|
5,342
|
3
|
3
|
5
|
Результаты расчета энтропии 2,2,4-триметилпентана представлены в табл. 2.12. Учитывая большое количество волчков в молекуле 2,2,4-триметилпентана и объемность некоторых из них, абсолютные значения энтропии при различных температурах рассчитаны без учета вклада на смешение конформеров. Для сравнения в табл. 2.11 приведены значения энтропии 2,2,4-триметилпентана, рекомендованные [1]. Ошибка расчета по методу статистической термодинамики возрастает с увеличением температуры от 298 до 600 К с -0,04 до -0,14 % отн. Зависимость  от температуры для состояния идеального газа представлена на рис. 2.11. от температуры для состояния идеального газа представлена на рис. 2.11.

Рис. 2.11. Зависимость энтропии 2,2,4-триметилпентана от температуры
Таблица 2.12
Расчет энтропии 2,2,4-триметилпентана методом статистической термодинамики
| T, K
|
Вклады для расчета энтропии, Дж/(моль·К)
|
 , расч., , расч.,
Дж/(моль·К)
|
, [1],
Дж/(моль·К)
|
| -Rln(σ)
|

|
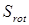
|
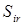
|
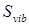
|
| 298
|
0,00
|
167,83
|
120,66
|
92,14
|
42,40
|
423,03
|
423,21
|
| 300
|
0,00
|
167,97
|
120,75
|
92,51
|
42,99
|
424,22
|
424,38
|
| 400
|
0,00
|
173,95
|
124,33
|
110,69
|
75,91
|
484,87
|
485,97
|
| 500
|
0,00
|
178,59
|
127,12
|
124,73
|
113,11
|
543,55
|
544,59
|
| 600
|
0,00
|
182,38
|
129,39
|
135,59
|
152,01
|
599,37
|
600,24
|
Пример 2.7
Методом статистической термодинамики рассчитать 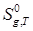 п-трет-бутилфенола при температурах 298, 300, 400, 500 и 600 К. п-трет-бутилфенола при температурах 298, 300, 400, 500 и 600 К.
Решение
1. Структурная формула молекулы рассматриваемого вещества такова:
2.

3. Вклад на симметрию  молекулы рассчитывается исходя из симметрии наружного вращения п-третбутилфенола, равной 1, и симметрии п-фениленового фрагмента, имеющего ось симметрии второго порядка: –R×ln(1·2). молекулы рассчитывается исходя из симметрии наружного вращения п-третбутилфенола, равной 1, и симметрии п-фениленового фрагмента, имеющего ось симметрии второго порядка: –R×ln(1·2).
4. Расчет энтропии выполнен аналогично примеру 2.6.; результаты приведены в табл. 2.13 и 2.14.
Дополнительно следует добавить только то, что при расчете вкладов на внутреннее вращение фрагментов не учитывается симметрия фениленового фрагмента (C6
H4
), где σ=2. В связи с этим следует включить σ=2 в расчет числа симметрии наружного вращения. Таким же образом следует поступать и с другими фрагментами, не подвергающимися учету во вкладе внутреннего вращения в энтропию.
Таблица 2.13
Молекулярные данные п-третбутилфенола
| Волчок
|
IAIBIC·10112 ,
г3/см6
|
Vr, Дж/моль
|
Ir·1040, г/см2
|
nmax
|
σ
|
ν
|
| t-Bu
|
6,505058
|
2415,098
|
81,83
|
6
|
3
|
1
|
| Me1(t-Bu)
|
21580,14
|
5,351
|
3
|
3
|
3
|
| Me2(t-Bu)
|
17338,03
|
5,359
|
3
|
3
|
4
|
| Me3(t-Bu)
|
16584,91
|
5,353
|
3
|
3
|
6
|
| OH
|
16668,59
|
1,348
|
2
|
1
|
7
|
Таблица 2.14
Расчет энтропии п-третбутилфенола методом статистической
термодинамики
| T, K
|
Вклады для расчета энтропии, Дж/(моль·К)
|
, расч.,
Дж/(моль·К)
|
| -Rln(σ)
|
|
|

|
|
| 298
|
-5,76
|
171,25
|
126,56
|
60,80
|
74,00
|
426,84
|
| 300
|
-5,76
|
171,38
|
126,64
|
61,07
|
74,79
|
428,12
|
| 400
|
-5,76
|
177,36
|
130,23
|
72,42
|
116,42
|
490,67
|
| 500
|
-5,76
|
182,00
|
133,01
|
81,64
|
160,31
|
551,21
|
| 600
|
-5,76
|
185,08
|
135,29
|
89,05
|
204,33
|
607,98
|
|