| Квалификационная работа
Разработка методики определения ультрамикрограммовых количеств
тяжелых металлов методом инверсионной вольтамперометрии
Содержание
Введение
Глава 1. Анализ следовых количеств веществ и электрохимические инверсионные методы
1.1 Анализ следовых количеств веществ и проблемы, стоящие перед соответствующими методами анализа
1.2 Электрохимические методы анализа следовых количеств веществ
1.3 Основы электрохимических инверсионных методов
1.4 Реакции, используемые для электролитического накопления
1.5 Типы рабочих электродов
1.6 Методы исследования процесса растворения
1.7 Избирательность определения
1.8 Роль предварительного отделения в инверсионных электрохимических определениях
1.9 Состояние и перспективы метода
1.10 Примеры практических приложений инверсионных методов
Глава II. Методы исследования и методика проведения эксперимента
2.1 Инверсионная вольтамперометрия
2.1.1 Как можно сконцентрировать определяемый микроэлемент на индикаторном электроде
2.2 Методика выполнения измерений массовой концентрации ионов кадмия, свинца, меди и цинка в питьевых, природных и очищенных сточных водах методом инверсионной вольтамперометрии
2.2.1 Назначение и область применения методики
2.2.2 Характеристика погрешности измерений
2.2.3 Метод измерений
2.2.4 Средства измерений, вспомогательные устройства, реактивы и материалы
2.2.5 Условия безопасного проведения работ
2.2.6 Условия выполнения измерений
2.2.7 Подготовка к выполнению измерений
2.2.8 Выполнение измерений
2.2.9 Форма представления результатов измерений
2.2.10 Контроль характеристик погрешности
2.3. Оборудование, применяемое в работе
Глава III. Экспериментальная часть
3.1 Порядок работы
3.2 Электрохимические параметры выполнения измерений на СУ-электроде
3.3 Выполнение измерений на углеродном электроде
3.3.1 Регистрация вольтамперограммы фонового раствора
3.3.2 Регистрация вольтамперограмм анализируемого раствора пробы с добавкой стандартного раствора ионов тяжелых металлов
3.4 Обсуждение результатов
Выводы
Список литературы
Приложения
Введение
Актуальность. Современный уровень развития технологии, биологии, медицины, охраны окружающей среды и других областей науки и техники выдвигает задачу определения малых количеств веществ во все более сложных объектах; поэтому требования, предъявляемые к методам анализа следовых количеств веществ, постоянно повышаются. Наряду с другими методами при анализе следовых количеств широко применяются электрохимические инверсионные методы, поскольку для очень многих элементов при относительно простом аппаратурном оформлении они приводят к хорошо воспроизводимым и правильным результатам.
В последние годы число публикаций, посвященных инверсионным методам, неуклонно растет; это связано с появлением новых приборов, а также с переходом к использованию ртутных пленочных и твердых электродов.
В опубликованных до сих пор обзорных работах и книгах по электрохимическому инверсионному анализу основное внимание уделялось работам с классическим ртутным электродом. Представляет определенный интерес рассмотреть возможность использования пленочных и твердых электродов.
В нашей работе мы постарались представить краткий обзор исследований с использованием методов инверсионного анализа в анализе объектов окружающей среды.
Таким образом, целью данной работы явилась разработка методики определения ультрамикрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии.
Реализация выдвинутой цели предопределила решение ряда частных задач:
1. анализ литературы по проблеме анализа микрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии;
2. анализ методических разработок по анализу различных объектов окружающей среды методом инверсионной вольтамперометрии;
3. выработка методики определения следовых количеств тяжелых металлов методом инверсионной вольтамперометрии, исходя из имеющихся материалов и аппаратуры;
4. апробирование данной методики на водных объектах;
5. анализ полученных данных.
Глава 1. Анализ следовых количеств веществ и электрохимические инверсионные методы
1.1 Анализ следовых количеств веществ и проблемы, стоящие перед соответствующими методами анализа
Для успешного анализа следовых количеств веществ необходимо решить три проблемы:
1) достаточно сильно снизить предел обнаружения, т. е. повысить величину отношения сигнал/шум (например, величину отношения электрического тока к остаточному);
2) достигнуть требуемой избирательности, т. е. возможности определять следовые количества элементов на фоне других, присутствующих в концентрациях, на несколько порядков более высоких; эту проблему обычно невозможно решить без использования предварительного отделения;
3) приготовить химические реактивы требуемой степени чистоты и усовершенствовать технику работы с очень разбавленными растворами, содержание растворенного вещества в которых уменьшается из-за адсорбции на стенках ячейки, гидролиза и т. д.
При определении очень малых количеств веществ наиболее часто используются радиохимические методы, особенно активационный анализ и методы радиоактивных индикаторов. Практически эти методы характеризуются наиболее высокой чувствительностью среди всех методов, применяемых при анализе следовых количеств; так, достигаемый в оптимальных случаях предел обнаружения равен ~10-21 г, что соответствует приблизительно десяти атомам или молекулам. Из физико-химических методов анализа к радиометрическим методам по пределам обнаружения приближаются флюориметрия в ультрафиолетовом свете (предел обнаружения 10-15 г) и эмиссионный спектральный анализ (предел обнаружения 10-12 г).
Некоторые физико-химические методы можно использовать для определений 10-6 – 10-10 г элемента (т. е. 10-6 – 10-10 моль/л). К таким методам относятся, например, спектрофотометрические (атомно-абсорбционная спектрофотомерия и флуориметрия), кинетические и каталитические, а также электрохимические (инверсионная вольтамперометрия) методы анализа.
Методы, основанные на взаимодействии электромагнитного излучения с веществом, бывают обычно более избирательны и имеют более широкую область применения; электрохимические инверсионные методы определения веществ характеризуются более низким пределом обнаружения, кроме того, они требуют более дешевой аппаратуры. В большинстве случаев электрохимические методы менее чувствительны к влиянию основы, чем оптические методы, при гораздо более высокой воспроизводимости и правильности результатов. Электроаналитические методы, в которых используется ртутный электрод, имеют более низкий предел обнаружения при определении некоторых металлов, например при определении Cd, Pb, Tl, Bi. [1]
1.2 Электрохимические методы анализа следовых количеств веществ
Классические электрохимические методы, в основе которых лежит стационарная поляризационная кривая (зависимость стационарного электрохимического тока от напряжения, наложенного на электродную систему), имеют предел обнаружения порядка микрограммов веществ (или ~10-5 моль/л). Такой предел обнаружения определяется отношением электролитического тока (определяемого электродным процессом изучаемого вещества) к остаточному току (сумма электролитических токов примесей, емкостного, обусловленного заряжением электрического двойного слоя тока, и «шума» измерительной цепи). Обычно величина остаточного тока составляет по меньшей мере 10-9 А, поэтому измеряемый электролитический ток должен быть достаточно большим для того, чтобы его можно было отличить от остаточного.
Предел обнаружения классических методов можно снизить, подавляя шум, используя более чувствительную измерительную аппаратуру, измеряя мгновенную концентрацию изучаемого вещества в диффузионном слое у электрода.
Для вращающихся и вибрирующих твердых электродов наряду с проблемами, связанными с достижением воспроизводимой конвективной диффузии, появляются трудности воспроизводимого обновления активной электродной поверхности. В настоящее время наиболее перспективными представляются вращающиеся дисковые электроды, для которых конвективный диффузионный поток электроактивного вещества может быть описан математически и активная поверхность которых с достаточно хорошей воспроизводимостью может быть обновлена полированием.
Чтобы снизить пределы обнаружения электроаналитических методов, необходимо предварительно сконцентрировать разбавленный раствор образца. Для этой цели применимы некоторые методы разделения (хроматография, жидкостная экстракция), при которых происходит отделение мешающих компонентов. Методики продолжительны и трудоемки, возможны потери части определяемого вещества в процессе концентрирования и введение загрязнений в анализируемую систему. Поэтому выгоднее проводить предварительное накопление в системе, в которой проводится измерение. Этот принцип положен в основу электрохимических инверсионных методов: определяемое вещество концентрируется электрохимически на индикаторном электроде (образуя амальгаму или пленку на поверхности электрода), а затем при обратном (электролитическом) процессе переводится в раствор. Таким образом исследуемое вещество в фазе электрода (на границе электрод — раствор) находится в существенно большей концентрации, чем первоначально, и чувствительность определения возрастает во много раз. [1]
1.3 Основы электрохимических инверсионных методов
Электролитическое накопление вещества из разбавленного раствора в большинстве случаев проводится при постоянном потенциале, который выбирается таким образом, чтобы требуемая электродная реакция протекала с достаточной скоростью. Раствор во время электролиза перемешивается, чтобы осуществлялся постоянный перенос деполяризатора из раствора. Для стационарных электродов по истечении определенного времени перемешивание прекращается и раствор успокаивается. За этот период поток вещества к электроду уменьшается, и соответственно величина электролитического тока также быстро падает до величины стационарного диффузионного тока. После стадии успокоения проводится растворение выделенного вещества. При исследовании зависимости тока от электродного потенциала, меняющегося линейно со временем, результирующая поляризационная кривая имеет вид пика, положение которого (потенциал полупика jр/2) характеризует данное вещество, а его высота (или площадь) пропорциональна концентрации вещества в растворе при поддержании постоянных условий предэлектролиза. Схема инверсионной вольтамперометрии приведена на рис. 1.1 и в табл. 1.1.
Таблица 1.1. Основная схема инверсионной вольтамперометрии*
| Стадия
|
| накопления
|
успокоения
|
растворения
|
| Наложенный потенциал
|
jel = const
|
jel = const
|
j = f(t)*
|
| Длительность
|
t
|
tr
|
—
|
| Ток
|
Поток
деполяризатора
|
I® IL
|
IS = f(j)
|
*Iel – электролитический ток, IL — предельный ток, IS — ток электролитического растворения.
Вольтамперные инверсионные методы называют катодными или анодными в зависимости от характера инверсионного процесса (восстановления или окисления соответственно).
Вообще в инверсионной вольтамперометрии нашли применение две методики работы. Согласно одной из них необходимо полное электролитическое выделение вещества из раствора и контроль тока в течение всего времени, необходимого для полного растворения осажденного вещества. В благоприятных условиях методика позволяет получать правильные и очень хорошо воспроизводимые результаты, хотя длительность определения, особенно при больших объемах раствора, является ее недостатком. При работе с очень малыми объемами образца она все же удобна, так как деполяризатор выделяется из раствора за весьма небольшой промежуток времени [2].
Чаще используется другая методика: накопление проводится в течение определенного времени при воспроизводимых условиях. В этом случае количество осаждаемого на электроде вещества является воспроизводимой долей общего количества вещества в исходном растворе. Методика требует сохранения постоянной скорости переноса вещества к электроду и удобна в тех случаях, если можно подобрать условия предварительного электролиза, чтобы осаждаемая доля составляла только 2 – 3 % от общего количества вещества.
Высота пика растворения обычно зависит от следующих факторов:
а) количества вещества, осажденного на электроде, которое является функцией его концентрации в растворе, потенциала накопления, продолжительности накопления, скорости потока вещества из объема раствора к электроду (т. е. интенсивности перемешивания или скорости вращения электрода), площади активной поверхности электрода, состава раствора, температуры и электрохимических свойств системы;
б) условий процесса растворения, особенно от скорости поляризации, площади активной поверхности электрода, скорости отвода продуктов от электрода.
Если суммарный электродный процесс включает и химическую реакцию, то на высоту пика оказывают влияние скорость этой реакции, характер продуктов реакции, растворимость образующихся соединений и т. п.
1.4 Реакции, используемые для электролитического накопления
Для накопления вещества могут быть использованы различные электрохимические и химические реакции, например: восстановление катионов до соответствующего металла, образование амальгамы или малорастворимого соединения, адсорбция. Для определения веществ различных классов существуют определенные типы реакций и в соответствии с природой образующегося осадка подбираются остальные условия, и, прежде всего рабочий электрод. Чаще всего используются перечисленные ниже типы реакций.
2. Металлы, способные образовывать достаточно концентрированные амальгамы, могут быть сконцентрированы на стационарном ртутном электроде. Металл, образовавшийся из ионов при их электровосстановлении, растворяется в ртутном электроде; затем он анодно растворяется из амальгамы; регистрируется анодный ток.
3. Ионы металла могут быть восстановлены до металла и накоплены на подходящем инертном электроде (благородный металл, графит) в виде пленки. Этот процесс наиболее часто используется для определения ртути, благородных металлов и металлов, не образующих амальгам. Определяемое вещество можно сконцентрировать на электроде в виде малорастворимого соединения. По способу образования последнего реакции подразделяют еще на два типа:
а) Малорастворимое соединение образуется при взаимодействии с ионами материала электрода. Такое соединение концентрируется на электроде при потенциале, соответствующем окислению электродного материала, затем регистрируется катодное растворение пленки (например, определение хлорид-иона на ртутном, или серебряном электроде). Подобные реакции можно использовать для косвенного определения некоторых металлов. Косвенное определение серебра или золота с применением стационарного электрода можно проводить в растворе, содержащем сульфид-ион. После добавления ионов серебра или золота регистрируемый первоначальный пик, соответствующий катодному растворению сульфида, уменьшается, так как часть сульфид-иона осаждается в виде малорастворимого сульфида серебра или золота.
б) Малорастворимое соединение образуется в виде пленки на электроде при взаимодействии с некоторыми компонентами основного электролита или с реагентом, добавляемым в раствор. Во время электродной реакции ионы определяемого вещества восстанавливаются или окисляются до степени окисления, в которой они участвуют в химической реакции, приводящей к образованию осадка. Таким образом, в химической реакции с реагентом принимают участие только ионы, полученные в результате электродной реакции; кроме того, образование осадка должно происходить быстрее, чем перенос продукта электродной реакции от электрода в объем раствора. Только при этих условиях количество образовавшегося осадка пропорционально концентрации вещества в растворе. Самым простым примером применения этой реакции является определение некоторых элементов, образующих малорастворимые оксиды, например определение марганца или свинца после окисления ионов Мn2+ и Рb2+ до МnО2 и РbО2.
4. Для предварительного концентрирования некоторых ионов можно использовать поверхностно-активные вещества. Эти вещества могут адсорбироваться на поверхности электрода и реагировать с определяемым ионом с образованием комплекса (тип 4а), или комплекс иона с поверхностным веществом образуется в растворе и затем адсорбируется на поверхности электрода (тип 4б). Электродная реакция при растворении сводится к восстановлению или окислению адсорбированного на электроде комплекса. Важно, что при этом типе реакций можно достичь накопления даже в отсутствие электролитического тока, хотя количество адсорбированного вещества часто зависит от наложенного на электрод потенциала.
Кроме этих четырех основных типов описаны различные косвенные определения, основанные, например, на вытеснении электроактивного металла из его комплекса электронеактивным металлом или на реакциях осаждения (примеры приведены при рассмотрении третьего способа).
Таблица 1.2. Реакции, используемые в методе инверсионной вольтамперометрии
| Тип реакции
|
Вещество в растворе
|
ПРЕДВАРИТЕЛЬНОЕ НАКОПЛЕНИЕ
|
РАСТВОРЕНИЕ
|
| 1
|
Mn+
|

|

|
| 2
|
Mn+
|

|

|
| 3а
|
An-
|

|

|
| 3б
|
Mn+
|
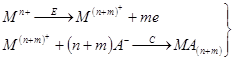
|

|
| 4а
|
Mn+
|
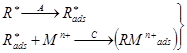
|

|
| 4б
|
Mn+
|

|

|
| 5
|
Mn+
|

|

|
| ·
|
An-
|


|

|
| 6
|
Mn+
|

|

|
*  - материал электрода; А – адсорбция; * - поверхностно-активные вещества; Е – электродная реакция; Ех – экстракция; С – химическая реакция; I – ионный обмен; индексы: w - водный; org – органический; ads – адсорбированный; ion – сорбированный на ионообменнике. - материал электрода; А – адсорбция; * - поверхностно-активные вещества; Е – электродная реакция; Ех – экстракция; С – химическая реакция; I – ионный обмен; индексы: w - водный; org – органический; ads – адсорбированный; ion – сорбированный на ионообменнике.
Экстракция адсорбированного на поверхности электрода вещества в малый объем растворителя.
Концентрирование на ионообменной мембране, размещенной непосредственно на поверхности электрода.
В табл. 1.2 кратко приведены рассмотренные выше реакции. Уравнения представляют собой только схемы процессов.
Предел обнаружения инверсионных методов можно улучшить при использовании каталитических реакций. В этом случае соединение, электродная реакция которого катализируется накопленным веществом, находится в растворе, и его каталитический ток измеряется в стадии растворения [3].
1.5 Типы рабочих электродов
Для понижения предела обнаружения, улучшения правильности и воспроизводимости большое значение имеет выбор рабочего электрода. Используемые электроды можно разделить на две группы: ртутные и твердые.
Самым простым, наиболее часто употребляемым типом ртутного электрода является стационарный ртутный капельный электрод. Такой электрод состоит из капилляра, соединенного с резервуаром, ртутная капля выдавливается из капилляра и стабилизируется у его устья. В самых распространенных устройствах капилляр вертикален и капля «подвешена» на ртутном столбике. Капилляр можно также изогнуть устьем вверх («лежащая» капля); такая конструкция дает определенные преимущества, так как улучшается стабильность капли и появляется возможность использовать капилляры с меньшим внутренним диаметром, что предотвращает обратную диффузию металла в капилляр. Кроме нестабильности капли в обратной диффузии, существенным недостатком таких стационарных ртутных электродов является проникновение раствора внутрь капилляра.
Как правило, в стационарном ртутном капельном электроде ртутная капля либо механически «подвешивается», либо осаждается электролитически на небольшом контакте из инертного металла (серебра, платины или золота). Вся поверхность контакта должна быть покрыта ртутью, а материал контакта должен иметь минимальную растворимость в ртути. При конкретных определениях все же необходимо учитывать возможность образования интерметаллических соединений определяемого вещества, с материалом контакта, особенно при использовании контакта из золота.
В последнее время все чаще применяется ртутный пленочный электрод: пленка ртути электролитически осаждается на благородный металл либо на графит или углерод. При использовании таких электродов можно снизить предел обнаружения, ибо у таких электродов отношение активной поверхности к объему ртути значительно возрастает (толщина пленки часто соответствует лишь нескольким молекулярным слоям). Кроме того, увеличивается разрешающая способность, так как диффузия металла в фазе электрода весьма ограничена и пики растворения получаются значительно более узкими. Пленочные ртутные электроды могут быть вращающимися, например вращающийся дисковый электрод.
Воспроизводимость результатов при работе со стационарными ртутными электродами зависит от воспроизводимости размера капли (или воспроизводимости массы ртути на твердой подложке). На практике не сложно достигнуть воспроизводимости, поэтому работать с ртутными электродами обычно очень удобно. Если образуется жидкая амальгама, то можно предположить, что осажденное вещество имеет одинаковую активность во всем объеме электрода. При анализе большинства смесей металлы, находящиеся в сравнимых концентрациях, не оказывают взаимного влияния друг на друга. Высокое перенапряжение выделения водорода позволяет определять металлы, дающие волны восстановления при весьма отрицательных потенциалах. В нейтральных растворах рабочая область потенциалов находится в интервале от —2,5 до +0,2 В.
Твердые электроды из платины, серебра, золота или графита используются при работе в области положительных потенциалов, где наблюдается растворение ртути.
При изготовлении электродов обычные графитовые материалы необходимо импрегнировать соответствующим способом (чтобы не происходило проникновения раствора в поры) или использовать угольные пасты. Пиролитический графит и стеклоуглерод не требуют импрегнирования.
При электролизе с твердыми электродами пленка образуется на поверхности электрода; это приводит к возникновению более сложной ситуации, чем в случае амальгамных электродов. Образование и растворение поверхностных пленок подчиняется более сложным зависимостям; на них оказывают влияние, например, структура и поверхностная энергия электрода, поверхностные каталитические явления, структура образовавшегося осадка. При анализе смесей часто наблюдаются взаимные помехи, и практически достигаемые пределы обнаружения часто гораздо выше, чем с применением ртутных электродов. Для получения воспроизводимых результатов необходимо, чтобы площадь активной поверхности электрода была постоянна и воспроизводимо обновлялась. Эта проблема решается специфично для каждого случая. Поверхностный слой оксидов на металлических электродах, например платиновом, может оказывать значительное влияние на электродную реакцию. Этот эффект следует изучить прежде, чем приступить к собственно определению. Несмотря на отмеченные недостатки, твердые электроды существенно расширяют возможности инверсионных методов и в настоящее время интенсивно изучаются.
Угольный пастовый электрод является особым типом твердого электрода. При соответствующей конструкции электрода можно выдавливать угольную пасту подобно тому, как выдавливается ртутная капля у стационарных ртутных электродов. Этим способом можно относительно легко обновлять активную поверхность, добиваясь достаточной воспроизводимости. Для приготовления угольной пасты используется подходящий органический растворитель или силиконовое масло. В некоторых случаях эти вещества могут, принимать участие в реакциях, которые происходят при предварительном концентрировании определяемых соединений. Например, при определении бромидов или броматов можно электролитически получить бром, который легко растворяется (и концентрируется) в растворителе, входящем в угольную пасту, но затем трудно добиться его количественного растворения из электрода. [1]
1.6 Методы исследования процесса растворения
Принцип накопления вещества и последующего его электрохимического растворения не является новым; он был, например, использован для измерения толщины металлических пленок. Збинден [4] уже в 1931 г. определял следовые количества меди, осаждая ее на платиновом электроде и измеряя зависимость анодного тока от времени при соответствующем постоянном потенциале в процессе растворения пленки металла.
В пятидесятых годах прием электролитического накопления и последующего электрохимического растворения вещества был распространен на многие электрохимические методы. Наибольшую известность получила вольтамперометрия с линейным изменением потенциала во времени [5 – 7] в связи с ее методической и инструментальной простотой.
Осциллографическая полярография [8] (рабочий электрод поляризуется переменным током с постоянной плотностью, амплитудой и частотой, а на экране осциллографа регистрируется функция dj/dt = f(j), квадратноволновая полярография [9] и переменнотоковая полярография [10], хронопотенциометрия и кулонометрия [11 – 13] могут быть также использованы для исследования процесса растворения. В некоторых случаях для повышения чувствительности определения применяют нестационарные методы. Для исследования процесса электрохимического растворения используются, таким образом, любые методы, основанные на изучении стационарных и нестационарных поляризационных кривых (табл. 1.3).
Таблица 1.3. Методы, применяемые при исследовании инверсионного процесса 3
| Контролируемый параметр
|
Измеряемая функция
|
Название метода
|
| Стационарные методы
|
| j
|
I = f(j)
|
Вольтамперометрия при постоянном потенциале
|
| j
|

|
Кулонометрия при постоянном потенциале
|
| j
|
Q = f(c)
|
Полярографическая кулонометрия
|
| I
|
Q = It
|
Кулонометрия при постоянном токе
|
| Нестационарные (потенциостатические) методы
|
| j
|
I = f(t)
|
Хроноамперометрия
|
| j = ji + wt
|
I = f(j)
|
Полярография и вольтамперометрия с переменным потенциалом (single sweep, multi-sweep)
|
| j + j(t)
|
I(t) = f(j)
|
Полярография и вольтамперометрия с наложением переменного напряжения (переменнотоковая полярография квадратноволновая полярография, импульсная полярография)
|
| Нестационарные (гальваностатические) методы
|
| I
|
j = f(t)
|
Хронопотенциометрия
|
| I + I sinwt
|

|
Осциллографическая полярография с переменным током
|
1.7 Избирательность определения
Рабочая область потенциалов для инверсионных электрохимических методов в водной среде находится в интервале от +1,5 до —2,5 В (от +0,2 до —2,5 В для ртутных электродов и от +1,5 до —0,7 В для графитовых электродов). В некомплексообразующих основных электролитах потенциалы пиков ряда элементов перекрываются или даже совпадают. Только в единичных случаях отличие в потенциалах пиков такое большое, что эти пики не оказывают друг на друга взаимного влияния. Относительно легко определить несколько металлов, если они находятся в растворе в одинаковых концентрациях. На практике, однако, часто требуется определить следовые количества одного вещества в присутствии большого избытка другого мешающего вещества, поэтому необходимо предварительно устранить его влияние.
Этого можно достигнуть, предварительно отделив мешающее вещество. Такой прием наиболее надежен, но на практике его применяют только в том случае, если для решения данной проблемы невозможно использовать другой, менее трудоемкий метод. Нежелательное влияние посторонних компонентов системы можно уменьшить и с помощью электрохимических способов: повышением избирательности накопления (например, применение потенциостата при определении металла в присутствии более электроотрицательного металла), выбором более селективного метода контроля процесса растворения (например, применение вольтамперометрии с переменной составляющей напряжения вместо классической вольтамперометрии или гальваностатического метода) или соответствующим подбором материала электрода.
В некоторых случаях можно сравнительно просто повысить избирательность при замене электролита: после стадии накопления выделенное вещество растворяется в чистом основном электролите или в другом пригодном растворе. Эти приемы до недавних пор были мало распространены, однако в настоящее время их применение расширяется [14], особенно в присутствии подходящих комплексообразующих реагентов [15].
1.8 Роль предварительного отделения в инверсионных электрохимических определениях
В ряде случаев, если определяемый компонент содержится в следовых концентрациях в очень сложных системах или же находится в присутствии очень большого избытка другого компонента (например, при анализе чистых реактивов, металлов и т. д.), предварительное отделение неизбежно. Для этой цели, как правило, применяют экстракционные и адсорбционные методы. Выбирая тот или иной метод для отделения, необходимо обращать особое внимание на то, чтобы компоненты системы не оказывали неблагоприятного влияния на исследуемую электролитическую реакцию (например, вследствие адсорбции поверхностью электрода избытка органических растворителей, или из-за появления следовых количеств поверхностно-активных веществ, выщелоченных из ионообменников, или в результате электрохимической реакции введенных реактивов и т. д.).
При экстракционном разделении обычно проводят реэкстракцию из органической фазы в водную или же органическая фаза минерализуется и растворяется в водном растворе перед непосредственным определением. Лишь в редких случаях, в основном при определении некоторых металлов, электролиз проводится прямо в неводной среде, в которой металл присутствует в виде ионного ассоциата. Этот прием перспективный, так как позволяет уменьшить число операций. К сожалению, имеется очень мало сведений по полярографии и вольтамперометрии комплексов металлов в неводной среде. [1]
1.9 Состояние и перспективы метода
В настоящее время в подавляющем большинстве инверсионных определений применяются процессы, сопровождаемые образованием амальгам металлов и металлических пленок (табл. 1.2, реакции 1 и 2). Другие процессы используются значительно реже. Как вытекает из вышеизложенного, электрохимические инверсионные методы являются очень подходящими для определения некоторых тяжелых металлов (Bi, In, Си, Tl, Pb, Cd, Sn, Zn) на ртутных электродах и некоторых благородных металлов (Ag, Hg) на твердых электродах. С помощью таких методов можно успешно определять указанные элементы в сплавах, чистых реактивах, водах, некоторых биологических материалах (в сыворотке крови, моче) и в некоторых продуктах питания.
Эти методы находят широкое применение при контроле загрязнений воды и воздуха. Например, классическая инверсионная вольтамперометрия часто используется для определения различных металлов в пресной и морской воде, а инверсионная вольтамперометрия с ртутным пленочным электродом на импрегнированной графитовой подложке — для контроля загрязнений воздуха [16].
Можно предположить, что значение и применение электрохимических инверсионных методов будет расширяться, особенно в связи с нарастающей важностью проблемы контроля загрязнений окружающей среды. Дальнейшее развитие этих методов зависит от фундаментальных исследований, которые концентрируются в нескольких основных направлениях.
Для более широкого применения инверсионных методов в текущих (серийных) и контрольных анализах необходима их автоматизация.
Электрохимические инверсионные методы в принципе невозможно использовать для непрерывных определений из-за необходимости осуществления последовательных стадий накопления и растворения, но они пригодны для выполнения автоматических серийных анализов в течение определенных временных интервалов, если имеется подходящая программирующая аппаратура. Хорошими примерами являются приборы с программным управлением, в которых используется ртутный стационарный капельный электрод [17] и вращающийся ртутный пленочный электрод [18].
Применение твердых электродов, особенно графитовых (в форме вращающихся дисковых электродов) или угольных пастовых, весьма перспективно. Эти электроды дают правильные и воспроизводимые результаты. Дальнейшее изучение пленочных ртутных электродов (это относится, прежде всего, к вращающимся дисковым электродам с тонким слоем ртути, осажденной in situ на графитовой подложке) позволит лучше использовать преимущества как ртутных, так и твердых электродов.
Интересны также различные методы измерения, которые до настоящего времени применялись лишь изредка, например потенциостатические и гальваностатические нестационарные методы, а в определенных случаях и полярография с переменной составляющей напряжения. Дальнейшее расширение возможностей электрохимических инверсионных методов может быть достигнуто путем сочетания с современными методами разделения, основанными на применении различных комплексообразующих реагентов, а также разными другими способами. Темпы развития этих методов зависят от состояния всех направлений электрохимии, теоретические данные которой могут быть полезны для разработки аналитических методик. С этой точки зрения особое значение имеют изучение кинетики электродных процессов на твердых электродах, адсорбционных явлений, электродных реакций с участием комплексов, электрохимических процессов в неводных средах, а также прогресс в аналитическом приборостроении (например, создание, приборов, основанных на операционных усилителях), который расширит набор методов исследования стадии растворения и позволит их полнее автоматизировать [1, 19, 20].
1.10 Примеры практических приложений инверсионных методов
В настоящее время в литературе имеется много примеров практического приложения инверсионных методов. В силу их разнородности невозможно привести полный обзор. Приведем несколько примеров анализа различных материалов, которые позволят составить представление о различных способах подготовки образца и о методах предварительного отделения.
Авторы [1] предлагают определение свинца в геологических образцах. Описывается методика подготовки образцов (циркона, монацита, пирохлора, гранита и андезита). Анодное определение свинца включает его продувку азотом, затем проводится предварительный электролиз на висящем ртутном капельном электроде при —0,6 В в течение 1 мин при перемешивании. После стадии успокоения (30 с) потенциал изменяется до 0 В со скоростью 40 мВ/с и регистрируется I—j-кривая. Концентрация свинца определяется по градуировочной кривой. При определении 3-10-3 % Рb, как указывают авторы [1, 21], относительное стандартное отклонение составляет 10%. Разрешение пиков In и Cd и пика Рb хорошее, но определению мешают высокие концентрации Тl.
В работе [22] показана возможность определения примесей кадмия, индия и цинка в свинце методом инверсионной вольтамперометрии. 0,2 г образца металлического свинца в кварцевом стаканчике растворяется в 5 мл горячей 3 М HNO3; Рb удаляется путем электролиза раствора с платиновым сеточным анодом при i = 0,1 А/см2 в течение 1,5 ч. В течение этого времени без прерывания тока электроды 4 раза вынимаются из раствора и осажденный РbО2 растворяется в 6—10 мл HNO3, содержащей 0,1 мл Н2О2. Затем раствор выпаривается досуха, и к остатку добавляется 3 мл Н2О. Выпаривание повторяется еще 3 раза. Наконец, остаток растворяется в 3 мл основного электролита (0,01 М KCl). Если используется ртутный пленочный электрод, то в первую очередь определяют Zn (jel = - 1,6 В, tel зависит от концентрации Zn). Для лучшего разрешения пиков Cd и In добавляется капля 1%-ного раствора этилендиамина; электролиз проводится в течение 3 мин при —1,6 В и определяются Cd и In [jp(Zn) = - 1,1 В; jр(In) = - 0,75 В; jp(Cd) = - 0,65 В]. При содержании 10-6 - 10-8 г/л рассмотренных элементов погрешность составляет 10-20%.
При определении свинца, меди и кадмия в пробах загрязненного атмосферного воздуха авторами [16] использовалась следующая методика. Образцы поглощались на стандартных фильтрах (размер 20 ´ 25 см) из стеклянных волокон (такие фильтры обычно используются при анализе суспендированных частиц). Из сложенного фильтра вырезаются два одинаковых квадрата (площадью по 13 см2), они разрываются на части и помещаются в мерную колбу 25 мл с узким горлом. Органические вещества разлагаются при добавлении 4 мл НСlО4 и последующим нагревании до 300°С в течение 30 мин. После этого колба заполняется водой до метки и оставляется стоять на 1—2 сут. Тем же способом подготавливается и контрольный опыт (кусочки фильтра имеют ту же величину). Из колбы с образцом затем отбирается аликвотная часть, соответствующая содержанию примесей в ~0,3 м3 воздуха, и в ней при обычных условиях на ртутном пленочном электроде с графитовой подложкой, импрегнированной воском, определяются Рb, Сu и Cd. Вначале проводится анализ контрольных растворов и лишь после этого — анализ образца. Поправка контрольного опыта вычитается. Контроль определения осуществляется благодаря проведению анализа образца с одного фильтра в двух разных ячейках, а при повторении анализа используются другие части фильтра. Предел обнаружения названных металлов ~10-6 г/м3. Были проведены исследования содержания свинца в биологических объектах. Например, в работе [23] проведено исследование содержания свинца в крови. 1 мл пробы крови помещается в колбу Киельдаля объемом 100 мл и минерализуется 2,5 мл 20%-ной H2SO4 в HNO3. Спустя 10 мин температура повышается до тех пор, пока не начнут выделяться белые пары H2SO4, после чего смесь охлаждается. Последовательно добавляют 1 мл HNO3, 1 мл 10%-ной НСlО4, две порции по 1 мл НСl (1 : 1) и, наконец, 5 мл воды. После каждой добавки раствор вновь нагревается до появления белых паров H2SO4. После охлаждения к образцу добавляют 20 мл воды и колбу нагревают до тех пор, пока не растворится весь осадок. После охлаждения весь объем раствора переносится в электролизер и проводится накопление при потенциалах от —0,6 до - 0,8В в течение 10 мин. Свинец определяют осциллографической полярографией с переменным током. Определению не мешает железо в концентрациях, в которых оно обычно присутствует в крови (~500 мкг/мл).
В работе [24] исследована возможность инверсионно-вольтамперомет-рического определения ртути в воздухе. Предложен состав раствора, экспериментальная установка, позволяющие проводить экстракцию ртути из воздуха и ее последующее инверсионно-вольтамперометрическое определение в растворе того же состава. Исследования проводились на золотом электроде. Найдено, что оптимальным составом раствора, позволяющим полностью улавливать ртуть из воздуха в диапазоне концентраций 0-100 мкг/м3 является: 1М HClO4 + 0,1M HCl + 10-6M I2. Установлена линейная зависимость аналитического сигнала от времени продувки и скорости паров ртути через электрохимическую ячейку, получена зависимость содержания ртути в ячейке от температуры прокачиваемого воздуха. На примере диметилртути показана принципиальная возможность определения органических соединений ртути в растворе того же состава. Предложен способ градуировки измерительной установки с использованием паров диметилртути.
Поскольку водные растворы йода неустойчивы на воздухе, то йод предложено вводить в раствор двумя способами. Первый способ - в виде спиртового раствора непосредственно перед измерениями, второй - электрохимическим генерированием йода из раствора калий йода. [24]
Томские ученые в тезисах [25] отмечают, что для определение ртути в последние годы широко применяется метод инверсионной вольтамперометрии (ИВА), отличающийся низкими пределами обнаружения и простотой применяемой аппаратуры. В работе оптимизированы условия получения сигнала ртути (0,0002-0,05 мг/л) методом ИВА в присутствии мешающих компонентов, таких как медь и железо, на тонкопленочном золотом электроде (in situ): фон – 0,1 М HNO3 c добавкой 4×10-6 M ионов Au (3+) и 0,005 М Cl-, Еэ = 0,2 В. Предложена пробоподготовка пищевых продуктов, сочетающая химическое окисление матрицы смесью концентрированных HNO3 и H2O2 с последующей обработкой раствора УФ-светом. Условия выбраны методом дробного факторного планирования эксперимента. Показано, что наиболее значимыми являются объем перекиси водорода и время химической минерализации. Для ряда матриц (молочные продукты, овощи) достаточна одна химическая стадия, при условии, что остаточная концентрация перекиси водорода в пробе менее 0,06%. Показано, что применение персульфата калия вместо перекиси водорода не эффективно в условиях пробоподготовки (удлиняется время и полнота фотоокисления). Для разложения проб зерновых и бобовых культур предложено вместо азотной кислоты использовать разбавленную 1:1 серную, иначе происходят большие потери ртути на стадии химической минерализации.
Показано [25], что эксимерная XeBr-лампа является альтернативой ртутным кварцевым лампам для разрушения органических веществ в процессе пробоподготовки различных образцов. Разработана методика определения ртути в пищевых продуктах (напитки, овощи, фрукты, молоко, творог, фасоль и др.) отличающаяся упрощенной пробоподготовкой. Она требует минимума операций, материалов, реактивов и посуды, что приводит к уменьшению величины холостого опыта.
Авторами [26] определение марганца в белых и красных винах проводили методом инверсионной вольтамперометрии с помощью вольтамперометрического анализатора “ИВА-5” (НПВП “ИВА”, г. Екатеринбург). Использовали трехэлектродную ячейку. В качестве рабочего электрода применяли толстопленочный графитсодержащий электрод (НПВП “ИВА”, г. Екатеринбург). Вспомогательным электродом служил стеклоуглеродный стержень; электродом сравнения – хлоридсеребряный электрод. Фоновым раствором служил аммиачно-хлоридный буферный раствор (рН 9,2 ± 0,2). рН раствора контролировали с помощью рН-метра-милливольтметра типа рН-150. Подготовку проб вина осуществляли на установке Digesdahl Digestion Apparatus Model 23130-20,21 (Hach Company, USA) путем мокрого его озоления с помощью концентрированной серной кислоты и пероксида водорода.
В работе выбраны оптимальные условия определения марганца в винах. Установлено, что сухие белые вина можно анализировать без предварительного разложения, а для красных вин необходима пробоподготовка. В результате анализа вин установлено, что содержание марганца в сухих винах составляет 0,5 – 1,9 мг/л, причем в красных винах содержание марганца меньше, чем в белых. Правильность полученных результатов проверялась методом «введено-найдено».
Современные пути развития метода инверсионной вольтамперометрии направлены на автоматизацию процесса анализа, решение проблемы пробоподготовки и создание экологически безопасных индикаторных электродов (сенсоров), позволяющих заменить классический ртутный или ртутно-пленочный сенсор, не ограничивая возможности метода. Новые типы электрохимических сенсоров (толстопленочных электродов) изготовлены на основе углеродных композиционных материалов по screen-printed – технологии в двух вариантах: в виде отдельных стрип-электродов и долгоживущих гибких электродов с заменяемой поверхностью. Электрохимическая регенерация их поверхности осуществляется в процессе измерений в автоматическом режиме. [27]
Толстопленочные углеродсодержащие электроды, химически модифицированные различными соединениями предварительно и in situ используются для определения Cd, Pb, Cu, Zn, Sn, Hg, As, Ni, Co, Cr, Fe, Se, Mn, Mo методом инверсионной вольтамперометрии.
Как указывают авторы [27], новые сенсоры экологически безопасны, высокочувствительны, селективны, обеспечивают высокие метрологические характеристики результатов анализа и могут использоваться с лабораторными анализаторами (ИВА-5) или проточными автоматизированными системами (ИВА-7). Программное обеспечение вольтамперометрических ПК-совместимых анализаторов ИВА-5 и ИВА-7 управляет автоматически работой всех подсистем (измерениями, электрохимической пробоподготовкой; контрольно-исполнительными устройствами), обрабатывает результаты измерений, накапливает и сохраняет результаты измерений. Предложенный комплексный подход к созданию вольтамперометрических приборов, программ, сенсоров для решения проблем вольтамперометрического мониторинга окружающей среды позволяет обеспечить экспрессный аналитический контроль содержания токсичных элементов как в стационарных и передвижных лабораториях, так и в автоматических проточных системах.
В связи с очень высокой токсичностью соединений мышьяка, его содержание в объектах окружающей среды подлежит обязательному контролю. ПДК мышьяка в воде и напитках находится в пределах от 0,05 до 0,2 мг/кг, что требует применения достаточно чувствительных методов определения. Распространенной является методика фотометрического определения мышьяка в воде, основным недостатком которой является сложная предварительная подготовка пробы к анализу. При определении As(3+) в водах и биологических объектах успешно применяются полярографические методы. Для определения мышьяка предлагается как анодная, так и катодная вольтамперометрия с использованием различных электродов (ртутно-пленочных, графитовых и стеклоуглеродных), различных фоновых электролитов и вторых элементов. Для повышения чувствительности определения мышьяка методом инверсионной вольамперометрии его обычно концентрируют на золотых и золото-графитовых электродах [28]. Работа посвящена разработке методики анализа воды на содержание мышьяка методом инверсионной вольтамперометрии с использованием золото-стеклоуглеродного электрода(ЗСУЭ), полученного методом «in situ»,что удешевляет анализ. Определены условия инверсионно-вольтамперометрического анализа мышьяка с использованием ЗСУЭ: фоновый электролит - Трилон Б (0,02 моль/дм3), время электролиза 120 сек, потенциал электролиза = -1,0 В. Диапазон определяемых концентраций 0,006-0,05 мг/дм3.Методом «введено – найдено» оценены возможности предлагаемой методики. Отклонение от истинного, оцененного по t-критерию, не значимо. Воспроизводимость результатов в изучаемой области концентраций не ниже 90%. Рассчитан предел обнаружения 0,006 мг/дм3.
Авторами [28] проведен анализ проб речной воды на содержание мышьяка методом инверсионной вольтамперометрии с использование ЗСУЭ. Сравнение полученных результатов с данными фотометрического анализа по t-критерию свидетельствует о хорошем совпадении результатов. Разработанная методика экспресснее фотометрической, т.к. не требует предварительного концентрирования, и превосходит ее по чувствительности.
Анализ нефтепродуктов на содержание меди, свинца, висмута на уровне 10-8 – 10-7%, проводили в условиях эффекта амальгамы аммония (ЭАА) при повышенной температуре. Сущность ЭАА заключается в следующем. В процессе электролиза раствора соли аммония (при определенных условиях) ионы аммония восстанавливаются на ртутном капельном электроде, и продукт реакции, аммиак, диффундирует в объем электрода. По мере его накопления в электроде объем последнего значительно увеличивается, возрастает при прочих равных условиях и ток электролиза. ИВА-определение проводили в 0,5 М растворе NH4Cl (индифферентный электролит) при температуре 50оС. Предварительное озоление пробы нефтепродукта массой 1-2 г проводили в калориметрической бомбе в атмосфере кислорода. [29]
Безусловно, большой интерес представляет способ количественного определения микроэлементов в нефтях без озоления пробы. Исключение этой стадии позволяет сократить общую продолжительность анализа и улучшить воспроизводимость его результатов. При этом навеску пробы массой 1,5-2 г предварительно обрабатывали при энергичном перемешивании 10 мл смеси конц. серной кислоты (r =1,84 г/см3) и пероксида водорода в соотношении 1:1 при температуре 60оС.
Изучены условия использования комплексов кадмия с ЭДТА и НТА для косвенного инверсионно-вольтамперометрического определения кобальта и никеля в нефтях и нефтепродуктах. Косвенно определяемый элемент М1 (Со, Ni) вытесняет из внутрикомплексного соединения элемент М2 (Сd), сравнительно легко определяемый методом ИВА. Поскольку раздельное определение никеля и кобальта при соизмеримых концентрациях в растворе затруднено, предложены условия предварительного ионообменного разделения этих элементов на сильноосновном анионите из растворов хлороводородной кислоты. [29]
Как показано в тезисах [30], для определения содержания иода в водах использовали метод катодной инверсионной вольтамперометрии и ртутно-пленочные электроды (РПЭ) в качестве индикаторных. Для дезактивации мешающего влияния органических веществ и растворенного кислорода пробу подвергали ультрафиолетовому облучению на фоне муравьиной кислоты. При этом происходит одновременное восстановление иодат-ионов и органических форм иода до иодид-ионов. Данная методика имеет много общих черт с методикой определения концентрации цинка, кадмия, свинца и меди в воде методом анодной инверсионной вольтамперометрии на РПЭ с фотохимической подготовкой проб. Это позволило совместить определение тяжелых металлов и йода в водах. Для этого проводили накопление цинка, кадмия, свинца, меди и регистрировали анодную вольтамперограмму, после чего делали остановку потенциала для накопления иодид-ионов и регистрировали катодную вольтамперограмму. Концентрацию элементов в пробе определяли методом добавок. Прецизионность результатов анализа, полученных при совместном и раздельном определении тяжелых металлов и йода, не превышает 20 %. Равноценной заменой ртутно-пленочному электроду оказались электроды, модифицированные твердым раствором ртути в серебре. Модифицирование проводили путем последовательного электрохимического нанесения ртути и серебра на рабочую поверхность электрода. В качестве подложки использовали серебро и различные виды углеродсодержащих электродов. Выбраны оптимальные условия модифицирования и проведения измерений, позволяющие получать аналитические сигналы цинка, кадмия, свинца, меди и йода, по своим параметрам не уступающие сигналам, полученным на РПЭ. Количество удовлетворительных результатов анализа, получаемых с использованием одного серебряного модифицированного электрода без регенерации поверхности – не менее ста, тогда как поверхность модифицированного углеродсодержащего электрода необходимо обновлять после анализа не более десяти проб.
Среди микроэлементов, попадающих в почвы со сточными водами, газовыми выбросами и производственными отходами, ртуть представляет наибольшую опасность. Вследствие повышения фонового содержания ртути в биосфере, контроль за уровнем загрязненности почв ртутью является актуальной задачей. В работе [31] проводились исследования по определению ртути в почвах методом инверсионной вольтамперометрии с использованием анализатора вольтамперометрического ТА-4 (ООО «НПП Томьаналит»). Ртуть в почвах может находиться в виде различных соединений: молекулярных, комплексных, неорганических, а также металлорганических. Для перевода этих соединений в раствор в виде электрохимически активной формы ртути почву обрабатывали при нагревании или УЗ-воздействии различными окислителями: 1) HNO3; 2) HNO3 + H2O2; 3) H2SO4 + HNO3; 4) H2SO4 + HNO3 + (NH4)2S2O8; 5) HNO3 + HCl. Полученные кислотные вытяжки фильтровали и разбавляли в 25-50 раз обессоленной водой. Дополнительно вытяжки обрабатывали: 1) озоном; 2) кипятили с перекисью водорода; 3) подвергали микроволновому разложению. Анализировали как обработанные вытяжки, так и просто разбавленные. Определение ртути проводили на фоне серной кислоты и хлорида калия. Индикаторный электрод – углеродсодержащий, приготовленный из смеси сажи и полиэтилена по технологии «литье под давлением», с нанесенной пленкой золота на торец электрода. Проверку правильности результатов анализа проводили методом добавок. Результаты, полученные после озонирования и микроволной обработки, сравнимы с результатами анализа разбавленных вытяжек. Результаты анализа проб после кипячения с перекисью водорода занижены на 50-70 %.
Наиболее воспроизводимые сигналы, позволяющие получить достоверные результаты, были получены при анализе разбавленной кислотной вытяжки смесью H2SO4+HNO3+(NH4)2S2O8. УЗ-воздействие позволяет сократить процесс приготовления вытяжки в 15-20 раз. Однако, в почвенных вытяжках, обработанных ультразвуком, мешающее влияние ионов железа из-за их более высокой концентрации увеличивает погрешность определения ртути. [31]
Разработана методика определения мышьяка в пищевых продуктах и продовольственном сырье методом инверсионной вольтамперометрии на углеродсодержащих электродах, модифицированных золотом (ЗУЭ). Выбраны оптимальные условия модифицирования поверхности ЗУЭ и проведения измерений, позволяющие проводить анализ порядка трехсот проб без регенерации поверхности электрода. Мешающее влияние кислорода устраняли химическим способом, используя в качестве фонового электролита сульфит натрия. Мешающее влияние цинка, меди и железа снижали образованием прочных комплексов различного состава с edta4—анионом и выбором параметров регистрации вольтамперограмм. Применение дифференциально-импульсной развертки поляризующего напряжения позволяет уменьшить погрешность измерения концентрации мышьяка, благодаря упрощению обработки аналитического сигнала. Однако, количество проб, анализируемых с использованием этой формы развертки без регенерации поверхности ЗУЭ – не более двадцати. [32]
Летучесть мышьякорганических соединений и хлорида мышьяка (III), а также достаточно низкие содержания мышьяка в пищевых продуктах (порядка 10-2 мг/кг) делает пробоподготовку при определении мышьяка достаточно сложной и требующей особого внимания. Наиболее часто при разложении проб для определения мышьяка используют способ, сочетающий методы мокрой минерализации и сухого озоления с добавками нитрата или оксида магния. С целью уменьшения погрешности определения мышьяка в пищевых продуктах, исследовали возможные потери мышьяка и их причины на каждом из этапов минерализации проб. Восстановление As(V) в минерализате до электрохимически активного As(III) авторы проводили пиросульфитом натрия, вместо широко используемого сернокислого гидразина. Это позволило сократить время подготовки проб на 1,5 часа. Были выбраны температурный режим, количество и последовательность добавляемых реагентов, при которых погрешность определения мышьяка, связанная с минерализацией проб, не превышает 15 %. [32]
Глава II. Методы исследования и методика проведения эксперимента
2.1 Инверсионная вольтамперометрия
Инверсионная вольтамперометрия является одним из вариантов электрохимических методов анализа, основанных на предварительном концентрировании определяемого компонента. Предварительное концентрирование осуществляется за счет перевода определяемого компонента из большого объема раствора с малой концентрацией на поверхность или в малый объем электрода. Перевод определяемого компонента из раствора на поверхность или в объем электрода может быть осуществлен за счет протекания соответствующей электрохимической реакции или за счет процесса адсорбции. После накопления на поверхности или в объеме электрода определяемое вещество подвергается электрохимическому превращению (восстановлению или окислению), причем этот процесс можно проводить в разных режимах:
1) Потенциодинамическом (при линейном изменении потенциала электрода во времени - инверсионная вольтамперометрия). На записываемой при этом вольтамперограмме (кривой “ток - потенциал”) будет наблюдаться пик, высота которого определяется концентрацией накопленного на электроде вещества и, следовательно, является аналитическим сигналом. Для повышения чувствительности используются различные варианты переменнотоковой, дифференциально-импульсной, квадратно-волновой и других видов вольтамперометрии.
2) Гальваностатическом (электрохимическое превращение накопленного на электроде вещества при постоянном токе - инверсионная хронопотенциометрия). Из записываемой при этом хронопотенциограммы (кривой “потенциал - время”) определяется так называемое переходное время (время полного электрохимического превращения накопленного вещества), которое однозначно связано с количеством накопленного вещества и является, следовательно, аналитическим сигналом.
3) Потенциостатическом (при постоянном потенциале электрода – инверсионная хроноамперометрия и кулонометрия). В этом случае записывается кривая “ток - время”, на основании которой может быть определено количество электричества, пошедшее на электрохимическое превращение накопленного на электроде вещества и являющееся, следовательно, аналитическим сигналом. Если при потенциостатическом превращении накопленного на электроде вещества наблюдаемый ток практически не меняется во времени, то он также может служить аналитическим сигналом.
Наибольшее применение получил вариант инверсионной вольтамперометрии (переменнотоковой или дифференциально-импульсной).
Существенными преимуществами инверсионных электрохимических методов (ИЭАМ) перед другими методами определения следовых количеств неорганических и органических веществ в растворах являются:
возможность определения значительного числа химических элементов Периодической системы и многих органических веществ;
низкие пределы обнаружения, достигающие для некоторых элементов (Cd, Bi, Tl, Pb, Sb, Ni) и органических веществ уровня 10-9 - 10-10 М;
высокая селективность ИЭАМ и хорошие метрологические характеристики методик на их основе;
легкость компьютеризации и автоматизации аналитических определений;
относительная простота и сравнительная дешевизна приборов для ИЭАМ.
В качестве рабочих электродов в инверсионной вольтамперометрии чаще всего используют ртутный капельный электрод (висящая ртутная капля), ртутный пленочный электрод, платиновый, стеклоуглеродный электроды, электрод из графитовой пасты и другие. На ртутном электроде удобно проводить определение концентрации катионов некоторых металлов, которые обратимо восстанавливаются в определенной области потенциалов (от потенциала окисления ртути до потенциалов восстановления фоновых катионов) и образуют со ртутью амальгамы. К таким элементам относятся, в частности, Cu, Cd, Zn и Pb. Накопление определяемых компонентов на электроде проводят при постоянном значении потенциала электрода. Значение потенциала предварительного накопления должно быть таким, чтобы процесс электровосстановления протекал на предельном токе (Id). При этом с помощью перемешивания раствора поддерживают условия стационарной диффузии, т.е. постоянные гидродинамические условия. В результате этого количество восстановившегося за определенное время компонента оказывается прямо пропорционально его исходной концентрации в растворе. По окончанию стадии предварительного катодного накопления перед съемкой анодных вольтамперных кривых перемешивание раствора прекращают, и в течение 10 – 15 сек осуществляют стадию успокоения раствора. Процесс окисления образовавшейся амальгамы проводят в неперемешиваемом электролите в потенциодинамическом режиме. Для увеличения чувствительности метода при съемке вольтамперограмм используют дифференциальную импульсную квадратно-волновую методику, которая позволяет исключить влияние емкостного и фонового токов на получаемые зависимости.
2.1.1 Как можно сконцентрировать определяемый микроэлемент на индикаторном электроде
Известны два способа концентрирования определяемого элемента на поверхности электрода: электролитический и адсорбционный. Электролитическое предварительное концентрирование впервые было использовано в начале 60-х годов прошлого века для определения примесей тяжелых металлов в особо чистых реактивах и воде, необходимых для бурно развивающейся тогда радиоэлектронной промышленности.
Электролитическое предварительное накопление проводят на микроэлектроде (обычно это капля ртути, каким-то образом подвешенная в анализируемом растворе, но может быть и микроэлектрод из углеродного или другого инертного материала) при потенциале предельного тока определяемых ионов до металла при интенсивном перемешивании раствора. Если электрод ртутный, образуется амальгама
Men+ + ne + Hg ® Me(Hg).
По истечении заданного времени (в зависимости от концентрации это 60–300 с) мешалку выключают и в течение ~ 15 с позволяют раствору успокоиться. Затем включают развертку потенциала и регистрируют вольтамперограмму в интервале от потенциала электролиза до 0 В. На вольтамперограмме наблюдаются анодные пики, обусловленные окислением металлов из амальгамы
Me(Hg) − ne ® Men+ + Hg.
Если стандартные потенциалы пар Меn+/Ме различаются заметно, пики на вольтамперограмме хорошо разрешаются и можно определить до 4–5 элементов одновременно.
Анодная инверсионная вольтамперометрия успешно применяется для одновременного определения следовых количеств ионов металлов, обладающих достаточно высокой растворимостью в ртути. К ним в первую очередь относятся медь, свинец, кадмий и цинк.
Разработаны простые и надежные методики определения этих элементов в разнообразных природных (воды поверхностные, грунтовые, морские, почвы, растения), биологических объектах и пищевых продуктах с пределами обнаружения порядка 10−8 – 10−9 моль/л.
Адсорбционное концентрирование появилось значительно позднее. Его суть заключается в том, что определяемый элемент накапливают в виде комплекса с адсорбированным на поверхности электрода лигандом. Концентрирование проводят при потенциале максимальной адсорбции лиганда, который устанавливают в предварительных экспериментах, в течение строго контролируемого времени при энергичном перемешивании раствора магнитной мешалкой. После выключения мешалки и успокоения раствора регистрируют катодную вольтамперограмму, изменяя потенциал электрода к более отрицательным значениям. На вольтамперограмме наблюдается пик восстановления иона металла из комплекса, сконцентрированного на поверхности электрода, или реже пик восстановления лиганда из этого комплекса. При контролируемых условиях (состав фона, рН, потенциал и время адсорбции, площадь поверхности электрода, скорость перемешивания) высота пика линейно зависит от концентрации иона металла в растворе. Этот метод анализа получил название “адсорбционная катодная инверсионная вольтамперометрия” или просто “адсорбционная вольтамперометрия”. В последнее двадцатилетие это один из самых надежных и чувствительных методов определения следовых количеств элементов в разнообразных объектах, особенно в объектах окружающей среды. К числу наиболее часто применяемых лигандов для адсорбционного концентрирования относятся 8-оксихинолин, пирокатехин, диметилглиоксим. [33 – 37]
2.2 Методика выполнения измерений массовой концентрации ионов кадмия, свинца, меди и цинка в питьевых, природных и очищенных сточных водах методом инверсионной вольтамперометрии
При выполнении работы нами была взяты за основу несколько методик после их анализа. [1, 38 – 41]
2.2.1 Назначение и область применения методики
Методика предназначена для определения массовой концентрации кадмия, свинца, меди и цинка в питьевых, природных, морских и очищенных сточных водах методом инверсионной вольтамперометрии. Методика позволяет выполнять измерение массовой концентрации указанных ионов в одной пробе одновременно.
Диапазоны измерения массовой концентрации ионов в пробе, подготовленной к измерениям: Cd, Рb, Сu – от 0,001 до 1,0 мг/дм3, Zn – от 0,010 до 1,0 мг/дм3.
Содержание растворенных форм определяют в фильтрованной пробе; суммарное содержание – в нефильтрованной пробе; содержание нерастворенных форм рассчитывают по разности найденных значений.
Расширение диапазона измеряемых концентраций ионов кадмия, свинца, меди и цинка в пробах продукции может быть достигнуто за счет разбавления или концентрирования анализируемой пробы.
2.2.2 Характеристика погрешности
измерений
Методика обеспечивает получение результатов анализа массовой концентрации ионов кадмия, свинца, меди и цинка в пробах питьевой, природной, морской и очищенной сточной воды с погрешностью, не превышающей значений, приведенных в таблице 2.1 при доверительной вероятности Р=0,95. Нормы погрешности измерений соответствуют ГОСТ 27384-87.
Таблица 2.1
| Наименование иона
|
Диапазон массовой концентрации, мкг/дм3
|
Границы относительной погрешности, ±мкг/дм3
|
| Вода питьевая, природная и морская
|
| кадмий
|
1 – 1000
|
0,11Х + 0,28
|
| свинец
|
1 – 1000
|
0,12Х + 0,28
|
| медь
|
1 – 1000
|
0,10Х + 0,25
|
| цинк
|
10 – 1000
|
0,07Х + 1,30
|
| Вода очищенная сточная
|
| кадмий
|
1 – 1000
|
0,18Х + 0,32
|
| свинец
|
1 – 1000
|
0,17Х + 0,30
|
| медь
|
1 – 1000
|
0,15Х + 0,30
|
| цинк
|
10 – 1000
|
0,10Х + 1,50
|
2.2.3 Метод измерений
Метод основан на электрохимическом концентрировании кадмия, меди, свинца, цинка на поверхности стеклоуглеродного электрода и последующем электрохимическом растворении при заданном потенциале с регистрацией вольтамперограммы.
Значения массовой концентрации ионов кадмия, меди, свинца, цинка в пробах продукции определяют методом добавок: сравнением величин аналитических сигналов, полученных для растворов проб и тех же проб после прибавления стандартных растворов с известной концентрацией анализируемых ионов.
2.2.4 Средства измерений, вспомогательные устройства, реактивы и материалы
Средства измерений
Потенциостат ПИ-50-1 и AUTOLAB (анализатор вольтамперометрический АКВ по ТУ 4215-001-18294344).
Государственные стандартные образцы состава растворов ионов кадмия (ГСО 5222), свинца (ГСО 6077), меди (ГСО 6073), цинка (ГСО 6064). Массовая концентрация ионов металлов в стандартных образцах 1,0 мг/см3 и относительная погрешность концентрации не более 1%.
При отсутствии стандартных образцов допускается использование стандартных растворов, приготовленных согласно ГОСТ 4212.
Весы лабораторные общего назначения с метрологическими характеристиками по ГОСТ 24104 с наибольшим пределом взвешивания 200г.
Набор гирь Г-2-200, ГОСТ 7328.
Колбы мерные наливные 2-50-2, 2-100-2, 2-1000-2, ГОСТ 1770.
Цилиндры мерные 1-10, 1-25, 1-50, ГОСТ 1770.
Пробирки мерные вместимостью 20 см3, ГОСТ 1770.
Пипетки мерные 4(5)-2-1, 4(5)-2-2, 6(7)-2-5, 6-2-10, 3-2-20, 3-2-25 по ГОСТ 20292.
Дозаторы пипеточные ДП-1-20, ДП-1-1000.
Допускается использование иных средств измерений, с характеристиками не хуже указанных выше.
Вспомогательные устройства
Шкаф сушильный лабораторный с регулятором температуры 40-150°С по ТУ 16-531-639.
Электропечь сопротивления камерная, лабораторная, СНОЛ 1,6-2,5/11-И2 по ТУ 16-531.704 с регулятором температуры 150-500°.
Лампа инфракрасная мощностью 250 или 500 Вт.
Аппарат для приготовления бидистиллированной воды (стеклянный) АСД-4 по ГОСТ 15150, ТУ 25-1173.103-84.
Баня водяная лабораторная с электрическим обогревом.
Плитка электрическая с закрытой спиралью по ГОСТ 14919.
Колбы конические: КН-1-250-29/32 ТС по ГОСТ 25336.
Воронки фильтрующие: ВФ-1-32 ПОР 40 ТХС, В-25-38 ХС, В-36-50 ХС по ГОСТ 25336.
Чашки выпарительные вместимостью 20-50 см3 кварцевые по ГОСТ 19908 или фарфоровые лабораторные № 3-5 по ГОСТ 9147.
Стаканы термостойкие: В-1-5 ТС, В-1-150 ТС по ГОСТ 25336.
Палочки из стекла по ГОСТ 21400.
Штатив химический лабораторный по ТУ 64-1.707.
Щипцы тигельные ЩТ по ТУ 64-1.973.
Допускается использование иных вспомогательных устройств, с характеристиками не хуже указанных выше.
Реактивы
Кислота азотная, ГОСТ 11225 (d = 1,42 г/см3).
Калий (натрий) хлористый, ГОСТ 4234.
Спирт этиловый ректификованный, технический, ГОСТ 18300.
Кислота хлористоводородная, ГОСТ 14261 (d =1,19г/см3).
Ртуть (II) азотнокислая, одноводная, ГОСТ 4520.
Магния оксид, ГОСТ 4526.
Магний азотнокислый, 6-водный, ГОСТ 11088.
Калий азотнокислый, ГОСТ 4217.
Калий дихромат, ГОСТ 4220.
Кислота серная, ГОСТ 4204.
Вода дистиллированная, ГОСТ 6709, вода бидистиллированная по ТУ 6-09-2502-77.
"Хромовая смесь" для мытья лабораторной посуды.
Допускается использование иных реактивов с техническими характеристиками не хуже указанных выше.
Материалы
1. Фильтры обеззоленные, «синяя лента».
Допускается использование иных материалов, с характеристиками не хуже указанных выше.
2.2.5 Условия безопасного проведения работ
При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.4.021.
Содержание вредных веществ в воздухе рабочей зоны не должно превышать норм установленных ГОСТ 12.1.005.
Электробезопасность при работе с электроустановками по ГОСТ 12.2.007. Запрещается включать в сеть приборы и работать на них без заземления.
Помещение должно быть оборудовано приточно-вытяжной вентиляцией.
Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
Исполнители должны быть проинструктированы о мерах безопасности в соответствии с инструкциями, прилагаемыми к приборам.
2.2.6 Условия выполнения измерений
1. При подготовке к выполнению измерений и выполнении измерений соблюдают следующие условия:
-Температура окружающего воздуха, °С 25 ± 10
-Атмосферное давление, кПа 86 – 101 (760 ± 30 мм рт.ст.)
-Относительная влажность воздуха, % 65 ± 15
-Частота питающей сети, Гц 50 ± 0,5
-Напряжение питания в сети В 220 ± 10
2.2.7 Подготовка к выполнению измерений
При подготовке к выполнению измерений должны быть выполнены следующие работы: подготовка посуды, приготовление реактивов, настройка прибора.
Подготовка посуды к выполнению измерений
Новую и загрязненную стеклянную посуду для анализа промывают в растворе "хромовой смеси", затем многократно споласкивают водопроводной водой, тщательно промывают дистиллированной водой и трижды ополаскивают бидистиллированной водой. Непосредственно перед использованием посуду промывают 1М раствором азотной кислоты и тщательно споласкивают бидистиллированной водой.
Приготовление растворов
1. Приготовление основных растворов ионов кадмия, свинца, меди и цинка
1.1. Приготовление основного стандартного раствора свинца с концентрацией 100 мг/дм3.
Вскрывают стеклянную ампулу стандартного образца состава раствора иона свинца с концентрацией 1,0 мг/см3, выливают в сухой стакан; 5,0 см3 раствора с помощью пипетки вместимостью 5,0 см3 переносят в мерную колбу вместимостью 50 см3 и доводят до метки бидистиллированной водой.
1.2. Приготовление основного стандартного раствора кадмия с концентрацией 100 мг/дм3.
Вскрывают стеклянную ампулу стандартного образца состава раствора иона кадмия с концентрацией 1,0 мг/см3, выливают в сухой стакан; 5,0 см3 раствора с помощью пипетки вместимостью 5,0 см3 переносят в мерную колбу вместимостью 50 см3 и доводят до метки бидистиллированной водой.
1.3. Приготовление основного стандартного раствора цинка с концентрацией 100 мг/дм3.
Вскрывают стеклянную ампулу стандартного образца состава раствора иона цинка с концентрацией 1,0 мг/см3, выливают в сухой стакан; 5,0 см3 раствора с помощью пипетки вместимостью 5,0 см3, переносят в мерную колбу вместимостью 50 см3 и доводят до метки бидистиллированной водой.
1.4. Приготовление основного стандартного раствора меди с концентрацией 100 мг/дм3.
Вскрывают стеклянную ампулу стандартного образца состава раствора иона меди с концентрацией 1,0 мг/см3, выливают в сухой стакан; 5,0 см3 раствора с помощью пипетки вместимостью 5,0 см3, переносят в мерную колбу вместимостью 50 см3 и доводят до метки бидистиллированной водой.
2. Приготовление аттестованных растворов ионов кадмия, свинца, меди и цинка
Аттестованные растворы с концентрацией 1,0 и 10,0 мг/дм3 готовятся отдельно для каждого иона согласно МИ 2334 из основных растворов (1.1 – 1.4), разбавляя их дистиллированной водой.
Объем основных растворов и мерных колб, массовая концентрация исходных и приготовленных растворов ионов кадмия, свинца, меди и цинка и другие необходимые сведения приведены в табл. 2.2
Таблица 2.2
Приготовление аттестованных смесей (АС) растворов ионов кадмия, свинца, меди и цинка
| Концентрация исходного раствора для приготовления АС, мг/дм3
|
Объем исходного раствора для приготовления АС, см3
|
Объем мерной колбы, см3
|
Концентрация приготовленного раствора АС, мг/дм3
|
Срок
хранения
|
| 100
|
5
|
50
|
10,0
|
30 дней
|
| 100
|
1
|
100
|
1,0
|
Готовят
в день
определения
|
3. Приготовление вспомогательных растворов
3.1 Приготовление раствора азотной кислоты концентрации 1 М.
В мерную колбу вместимостью 1000 см3 помещают 200-300 см3 бидистиллированной воды и вносят цилиндром 70 см3 концентрированной азотной кислоты, перемешивают, охлаждают и доводят до метки бидистиллированной водой.
3.2. Приготовление раствора азотной кислоты концентрации 0,1М
В мерную колбу вместимостью 1000 см3 вносят пипеткой 7,0 см3 концентрированной азотной кислоты и доводят до метки бидистиллированной водой.
3.3. Приготовление раствора хлористоводородной кислоты концентрации 1М.
В мерную колбу вместимостью 1000 см3 помещают 200-300 см3 бидистиллированной воды и вносят цилиндром 80 см3 концентрированной хлористоводородной кислоты, перемешивают охлаждают и доводят до метки бидистиллированной водой.
3.4. Приготовление раствора ртути (II) азотнокислой концентрации 0,01М.
Навеску ртути (II) азотнокислой массой 0,343 г взвешивают на аналитических весах, помещают в мерную колбу вместимостью 100 см3 и растворяют в 50 см3 раствора азотной кислоты концентрации 0,1М и доводят до метки бидистиллированной водой. Раствор хранят в темноте.
3.5. Приготовление раствора фонового электролита.
В мерную колбу вместимостью 1000 см3 вносят 50 см3 раствора хлористоводородной кислоты концентрации 1М, 10 см3 раствора азотнокислой ртути концентрации 0,01М и доводят до метки бидистиллированной водой. Раствор хранят в темноте.
Все растворы хранят в посуде из боросиликатного стекла.
4. Установка, включение и подготовка прибора к выполнению измерений
Устанавливают прибор в соответствии с руководством по эксплуатации. Параметры работы прибора устанавливают в соответствии с Руководством по эксплуатации и согласно таблице 2.3
Заполняют хлорсеребряный электрод сравнения насыщенным раствором хлористого калия за 48 часов до начала работы.
Устанавливают электрохимическую ячейку в соответствии с руководством по эксплуатации анализатора.
Таблица 2.3
Параметры измерений при регистрации вольтамперограмм
| Наименование параметров
|
Режим
|
| Ячейка
|
3-х электродная
|
| Вид полярографии
|
Переменно-токовая инверсионная
|
| Потенциал накопления, В Zn, Cd, Pb, Cu
Cd, Pb, Cu
|
-1,3
-0,9
|
| Амплитуда развертки, В Zn, Cd, Pb, Cu
Cd, Pb, Cu
|
1,45 – 1,50
1,05 – 1,10
|
| Диапазон тока
|
в зависимости от концентрации определяемого иона в пробе
|
| Потенциал электрохимической очистки рабочего электрода, В
|
0,0
|
| Время электрохимической очистки электрода, сек.
|
равно времени накопления
|
| Время накопления, сек (в зависимости от содержания иона)
|
не менее 60
|
| Амплитуда переменного напряжения, мВ
|
от 3 до 30
|
| Скорость линейной развертки потенциала, мВ/сек
|
20-50
|
| Направление развертки
|
Анодное (положительное)
|
| Потенциал аналитического пика растворения, В (ориентировочное значение)
|
Zn Cd Pb Cu
(-1.0) (-0.6) (-0.4) (-0.05)
|
2.2.8 Выполнение измерений
1. Отбор и хранение проб. Отбор, консервация и хранение проб питьевой воды по ГОСТ 24481; отбор, консервация и хранение проб воды сточной по инструкции НВН 33.5.3.01. При отборе проб составляют акт отбора, в котором указывают:
цель анализа, предполагаемые загрязнители;
место, время отбора;
номер пробы;
должность, фамилия лица, отбирающего пробу;
дата.
Примечание: для каждого анализа отбирают по три параллельных пробы воды (одна резервная). Для доставки в лабораторию сосуды с пробами упаковывают в тару, обеспечивающую сохранность и предохраняющую от резких перепадов температуры. Вода не должна подвергаться воздействию прямого солнечного света.
2. Подготовка проб к анализу.
2.1. Измерения массовой концентрации ионов меди, свинца, кадмия и цинка выполняются в одной пробе. При проведении анализов одновременно готовят две параллельных пробы.
2.2. Подготовка проб питьевой и природной пресной воды.
2.3. Пробу подготовленного к испытанию образца воды объемом 100 см3 переносят в выпарительную чашку, добавляют 1–2 см3 концентрированной азотной кислоты. Содержимое чашки упаривают до «влажных солей». Если остаток темный, кислотную обработку повторяют до его осветления.
2.4. Если остаток не осветляется, пробу упаривают досуха и прокаливают в муфельной печи при 450°С в течение 30 мин.
2.5. В чашку с золой добавляют 1 см3 1М хлористоводородной кислоты и 5 см3 раствора фонового электролита. Если зола плохо растворяется, чашку подогревают на водяной бане.
2.6. Раствор пробы, подготовленной к выполнению измерений: раствор, полученный в результате полной минерализации пробы, охлаждают и количественно переносят в мерную пробирку вместимостью 20 см3 через бумажный фильтр, увлажненный раствором фонового электролита. Раствор доводят до метки фоновым электролитом.
3. Подготовка проб очищенной сточной воды.
Подготовку проб сточной воды выполняют по 2.1 – 2.6 с обязательным выполнением операций по 2.2 (прокаливанием остатка в муфельной печи при 450°С в течение 30 мин).
4. Раствор контрольной "холостой" пробы готовят к выполнению измерений аналогично пробам воды, используя вместо пробы бидистиллированную воду.
5. Выполнение измерений
Выполнение измерений массовой концентрации ионов меди, свинца, кадмия и цинка в подготовленных пробах включает следующие основные операции (табл. 2.4):
Таблица 2.4
| п.п. МВИ
|
Наименование основной операции выполнения измерений
|
Раствор пробы, подготовленной для измерения (№№ п.п. МВИ)
|
| 9.3.1.
|
регистрация вольтамперограмм "холостой" пробы
|
9.2.4
|
| 9.3.2.
|
регистрация вольтамперограмм проб
|
9.2.2.4, 9.2.3
|
| 9.3.3.
|
регистрация вольтамперограмм проб с добавками растворов соответствующих ионов
|
9.2.2.4,9.2.3 с добавками
|
| 9.3.4.
|
очистка электродов
|
Серия измерений пробы: (холостой опыт – проба – проба со всеми добавками) выполняется при параметрах измерений, установленных для каждого измеряемого иона.
6. Регистрация вольтамперограмм «холостой» пробы.
6.1. В стеклоуглеродный тигель электрохимической ячейки помещают 20 см3 «холостой» пробы.
6.2. Закрепляют тигель.
6.3. Устанавливают потенциал электрохимической очистки рабочего электрода в соответствии с таблицей 2.3 и в течение не менее 60 секунд не регистрируя вольтамперограммы производят электрохимическую очистку электрода. Выключают ячейку.
6.4. Устанавливают параметры измерений (потенциал накопления, амплитуду развертки, диапазон тока и время накопления) в соответствии с рекомендациями табл. 2.3. Включают ячейку, регистрируют вольтамперограммы и выключают ячейку.
Примечание: в случаях измерения в пробах воды массовой концентрации ионов меди, свинца и кадмия (без Zn) рекомендуется установить потенциал накопления (-0,9 В) и соответственно амплитуду развертки 1,05-1,10 В.
6.5. Устанавливают потенциал очистки электрода 0,0 В, включают ячейку, выполняют электрохимическую очистку электрода в течение времени, равном времени накопления и выключают ячейку.
Цикл операций повторяют не менее трех раз.
Электрохимическая очистка электрода обязательна после записи каждой вольтамперограммы.
6.6. Результаты измерений (регистрация вольтамперограмм раствора «холостой» пробы и условия выполнения всех измерений) записываются в программном интерфейсе системы сбора и обработки данных (самописец).
7. Регистрация вольтамперограмм раствора пробы.
7.1. В стеклоуглеродный тигель помещают 20 см3 раствора пробы, подготовленной к измерениям.
7.2. Закрепляют тигель.
7.3. Выполняют операции по п. 6 при значениях параметров измерений, установленных при регистрации вольтамперограмм «холостой» пробы.
Значения потенциалов пика окисления определяемых металлов являются качественной характеристикой элемента; их ориентировочные значения приведены в таблице 2.3.
8. Регистрация вольтамперограмм пробы с добавками.
8.1. Добавку растворов АС определяемых ионов дозатором пипеточным вносят в тигель с пробой после регистрации вольтамперограмм и очистки электрода.
8.2. Объем и концентрация добавляемых в ячейку растворов АС (табл. 2.2) устанавливаются экспериментально для каждой пробы таким образом, чтобы высота аналитического пика измеряемого иона при регистрации вольтамперограмм пробы с добавкой увеличилась в 1,5–3 раза при значениях параметров, установленных при регистрации вольтамперограмм пробы.
Суммарный объем всех растворов АС, добавленных в ячейку, не должен превышать 10% от объема пробы.
8.3. Регистрацию вольтамперограмм в серии измерений: «холостая» проба, проба и проба с добавками соответствующего иона выполняют при одинаковых параметрах измерений для каждого иона.
8.4. В первую очередь выполняют измерения массовой концентрации ионов свинца, кадмия и цинка. Для этого в пробу вводят добавки растворов соответствующих ионов (табл. 2.1) и выполняют операции по п.6.
8.5. Измерение массовой концентрации ионов меди в пробе с добавками обычно проводят после того, как выполнена регистрация вольтамперограмм выше.
9. Очистка электродов.
После проведения серии анализов или в конце работы ячейку и электроды тщательно промывают бидистиллированной водой, рабочие электроды механически очищают сухой фильтровальной бумагой, затем бумагой, смоченной этанолом, вспомогательный (хлорсеребряный) электрод ополаскивают раствором хлористоводородной кислоты, промывают бидистиллированной водой и помещают в насыщенный раствор хлористого калия.
10. Регистрация измерений
Результаты измерений выполненных серий регистрируются в соответствии с руководством по эксплуатации и могут быть выведены на дисплей или принтер, сформированы в отчеты и протоколы испытаний.
2.2.9 Форма представления результатов измерений
1. Результаты анализа регистрируются в виде протокола в программном интерфейсе системы сбора и обработки данных (выводятся на самописец).
2. Результаты измерений массовой концентрации ионов в воде представляют в виде:
(Хср.± D)мг/дм3
где: Xср - результат анализа пробы, мг/дм3
D – абсолютная погрешность измерений массовой концентрации ионов, мг/дм3, при Р = 0,95.
Значение в рассчитывают по формуле: в = в × Х i ср /100, мг/дм3.
где в - значение относительной погрешности измерения массовой концентрации ионов, %.
2.2.10 Контроль характеристик погрешности
1. Оперативный контроль воспроизводимости.
1.1. Образцами для контроля являются реальные пробы воды. Объем отобранной для контроля пробы должен соответствовать удвоенному объему, необходимому для проведения анализа по методике.
1.2. Отобранную пробу делят на две равные части, которые анализируют в точном соответствии с прописью методики, максимально варьируя условия проведения анализа, т.е. получают два результата анализа, используя разные партии реактивов и наборов мерной посуды. Оперативный контроль воспроизводимости проводят через каждые 20 проб.
Воспроизводимость результатов анализа признают удовлетворительной, если результат контрольных измерений DK не превышают норматив D:

где X1, Х2 – результаты анализа одной и той же пробы, полученные в условиях внутрилабораторной воспроизводимости, мг/дм3;
D – норматив оперативного контроля воспроизводимости, табл. 2.5.
1.3. При превышении норматива оперативного контроля воспроизводимости эксперимент повторяют. При повторном превышении норматива D, выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
2. Оперативный контроль погрешности методики
2.1. Оперативный контроль погрешности методом добавок
Образцами для контроля являются реальные пробы воды. При применении метода добавок объем отобранной для контроля пробы должен соответствовать удвоенному объему, необходимому для проведения анализов по методике. Отобранную пробу делят на две равные части, первую из которых анализируют в точном соответствии с прописью методики. Во вторую часть вводят добавку CD аттестованного раствора, а затем анализируют в точном соответствии с прописью методики. Добавка должна составлять (50-150)% от содержания определяемых ионов в пробе.
Точность контрольных измерений признают удовлетворительной, если выполняется условие:

где К – норматив оперативного контроля погрешности, мг/дм3.
При внешнем контроле (Р = 0,95): К = d, где в - граница погрешности по табл. 2.1. При внутрилабораторном контроле (Р = 0,90): К = 0,84 d.
Таблица 2.5
Значения нормативов оперативного контроля характеристик погрешности
| Наименование иона
|
Диапазон массовой концентрации, мкг/дм3
|
Характеристики погрешности, мкг/дм3
|
| D
|
К
|
D
|
d
|
| Вода питьевая, природная и морская
|
| кадмий
|
1-1000
|
0,11х+0,28
|
0,11x+0,28
|
0,15х+0,39
|
0,08x+0,20
|
| свинец
|
1-1000
|
0,12х+0,28
|
0,12x+0,28
|
0,17х+0,39
|
0,09x+0,20
|
| медь
|
1-1000
|
0,10X+0,25
|
0,10x+0,25
|
0,14x+0,35
|
0,07х+0,18
|
| цинк
|
10-1000
|
0,07x+1,30
|
0,07x+1,30
|
0,10x+1,8
|
0,05х+1,0
|
| Вода очищенная сточная
|
| кадмий
|
1-1000
|
0,18х+0,32
|
0,18х+0,32
|
0,25x+0,45
|
0,12x+0,22
|
| свинец
|
1-1000
|
0,17х+0,30
|
0,17x+0,30
|
0,24x+0,42
|
0,12x+0,21
|
| медь
|
1-1000
|
0,15x+0,30
|
0,15x+0,30
|
0,21х+0,42
|
0,11x+0,21
|
| цинк
|
10-1000
|
0,10x4-1,5
|
0,10x4-1,5
|
0,14х+2,1
|
0,07X41,0
|
Точность контрольных измерений признают удовлетворительной при выполнении условия:

где С и CD - результаты контрольных измерений содержания ионов в пробе и в пробе с добавкой; XD – значение добавки, мг/дм3
KD – норматив оперативного контроля погрешности, рассчитываемый по следующим формулам: при проведении внутрилабораторного контроля (Р=0,90): KD = 0,84 ÖDx2 + DxD2; при проведении внешнего контроля (Р = 0,95): KD = ÖDx2 + DxD2, где Dx и DxD –значения абсолютной погрешности результатов анализа пробы и пробы с добавкой, соответственно.
При превышении норматива оперативного контроля погрешности проводят повторное определение ионов в пробе. При повторном превышении указанного норматива К выясняют причины, приводящие к неудовлетворительным результатам, и устраняют их.
Метрологические характеристики приведены в виде зависимости от значения X – среднего арифметического результата параллельных определений. [1, 38 – 41]
2.3 Оборудование, применяемое в работе
Количественное определение ионов цинка, кадмия, свинца и меди методом ИВА проводили с помощью импульсного потенциостата ПИ-50-1 (стационарные I - j кривые регистрировались x - y потенциометрическим двухкоординатным самописцем ЛКД 4-003) и электрохимического комплекса AUTOLAB PGSTAT 30 (Голландия). В качестве индикаторного электрода использовались полупогруженные игольчатые стеклоуглеродные электроды. Электродом сравнения служил хлорсеребряный электрод. Анодом и одновременно контейнером для раствора служил стеклоуглеродный тигель. Потенциал электролиза j = -1,6 В, время накопления – 300 секунд, процесс электрорастворения элементов с поверхности индикаторного электрода и регистрация аналитических сигналов на вольтамперограмме проводился при линейно меняющемся потенциале от –1,2 до 0,05 В относительно хлорсеребряного электрода при заданной чувствительности прибора. Для удаления кислорода проводили облучение раствора УФ в течение 15-20 минут. Потенциалы максимумов регистрируемых анодных пиков Zn, Cd, Pb и Cu соответственно равны: -0,9В; -0,6В; -0,4В; -0,05В.
Пробы для анализа были взяты из различных источников (табл. 2.6)
Таблица 2.6
| № пробы
|
Вид
|
Местоположение забора пробы
|
| 1
|
Сточные воды
|
район «5 микрорайон»
|
| 2
|
район «Вольный Аул»
|
| 3
|
район «Искож»
|
| 1
|
Водопроводная вода
|
район КБГУ
|
| 2
|
район «Горный»
|
| 3
|
район «Искож»
|
Глава III. Экспериментальная часть
Измерения проводили по трехэлектродной схеме: рабочий электрод – стеклоуглеродный стержень (Æ 0,7 мм), вспомогательный электрод – стеклоуглеродный тигель (V = 25 см3) и электрод сравнения – хлорсеребряный электрод в насыщенном растворе KCl (х.с.э.).
В работе используют растворы:
фоновый раствор – 0,1М NaCl c pH 3;
исходные растворы металлов:
1) Cu, Cd и Zn по 10-1 г/л;
2) 10-1 г/л Pb при рН 3.
Электрохимическое накопление определяемых металлов на катоде проводили за счет электровосстановления при потенциале -1,1В (относ. х.с.э.). Съемку анодных вольтамперных кривых проводили в интервале потенциалов -1,1 - +0,1 В при скорости развертки потенциала 2 – 5 мВ/с.
Перед проведением измерений в течение 15 мин, а также при проведении накопления металла через раствор пропускали инертный газ (очищенный и осушенный аргон) или облучали УФ-излучением для удаления из раствора растворенного кислорода. Во время съемки вольтамперограмм инертный газ пропускали над раствором.
3.1 Порядок работы
В ячейку заливали 30 мл фонового раствора с неизвестной концентрацией катионов Cu, Cd, Zn и Pb. Пропускали через раствор в течение 15 мин инертный газ (или облучали УФ-светом), проводили накопление металлов в течение установленного времени и снимали анодную вольтамперную кривую.
Рассчитывали, какие добавки растворов определяемых металлов с концентрацией 10-1 г/л нужно внести в ячейку для изменения концентрации рабочего раствора на определенную величину. Вводили рассчитанные добавки, 10 мин пропускают через раствор инертный газ и снимали еще 3 вольтамперограммы при том же времени накопления.
Для каждого пика определяли средние значения высоты пика, полученные в исходном растворе и в растворе после введения добавки. Концентрации катионов в исходном растворе находили с помощью соотношения
Ip1/Ip2 = cx /(cx+c),
где
Ip1 - высота пика в исходном растворе;
Ip2 - высота пика после введения в раствор добавки;
cx - концентрация катиона данного металла в исходном растворе;
c - изменение концентрации соответствующего катиона в растворе в результате введения добавки.
Вводили в ячейку вторично такие же добавки катионов металлов, пропускали инертный газ и проводили аналогичные измерения. С помощью соотношения Ip1/Ip3 = cx/(cx+2c) (где Ip3 - высота пика после введения в раствор второй добавки) также определяют исходную концентрацию в растворе ионов исследуемых металлов.
Рассчитывали средние значения концентрации ионов Cu, Cd, Pb и Zn в исходном растворе, полученные в результате расчетов после введения первой и второй добавок.
Схему процесса можно представить таким образом.

3.2 Электрохимические параметры выполнения измерений на СУ-электроде
Таблица 3.1
Параметры выполнения измерений
| Наименование
параметра
|
Единицы
измерения
|
Величина
параметра
|
| Потенциал очистки электрода
|
мВ
|
-200
|
| Продолжительность очистки
|
с
|
60
|
| Потенциал накопления
|
мВ
|
800
|
| Продолжительность накопления*
|
с
|
180
|
| Мешалка
|
вкл.
|
| Продолжительность успокоения раствора
|
с
|
10
|
| Скорость развертки потенциала
|
мВ/c
|
150
|
| Начало развертки потенциала
|
мВ
|
800
|
| Конец развертки потенциала
|
мВ
|
-200
|
| Шкала измерения катодного тока
|
мкА
|
20мкА/2 мкА
|
| Электродная схема ячейки
|
–
|
трехэлектродная
|
| Вид полярографии
|
–
|
постояннотоковая
|
*При низкой интенсивности сигнала (менее 0,25 мкА) допускается увеличение времени накопления со 180 до 900 с включительно
3.3 Выполнение измерений на углеродном электроде
3.3.1 Регистрация вольтамперограммы фонового раствора
Фоновый раствор переносили в стеклянный стакан электрохимической ячейки и погружали в него электроды.
Запускали процесс измерений фона. По окончанию измерения на экране монитора выводилась вольтамперограмма фонового раствора. При соблюдении требований по квалификации реактивов и использовании чистой посуды, пипеток и электродов в фоновом растворе должны отсутствовать пики (рис. 3.1).
После того, как поверхность электрода была готова к работе, переходили к регистрации вольтамперограмм анализируемого раствора пробы.

Рис. 3.1. Вольтамперограммы фонового раствора при тестировании рабочего электрода: 1 – вольтамперограммы фонового раствора, означающие, что поверхность рабочего электрода не готова к работе, 2 - вольтамперограммы фонового раствора, полученные на рабочем электроде с подготовленной поверхностью. Можно переходить к измерениям.
Регистрацию вольтамперограмм повторяли до тех пор, пока относительная разность высот пиков в двух последних вольтамперограммах не будет превышать 5 – 8 % (обычно максимум до четырёх измерений при подготовленной поверхности электрода).
3.3.2 Регистрация вольтамперограмм анализируемого раствора пробы с добавкой стандартного раствора ионов тяжелых металлов
После выполнения регистрации вольтамперограммы анализируемого раствора пробы в стакан с анализируемым раствором вносили пипеткой добавку стандартного раствора ионов тяжелых металлов. Объем добавки (VД), который не должен превышать 2,0 см3, подбирали таким образом, чтобы после её внесения высота пика на вольтамперограмме увеличивалась в 1,5 – 2,0 раза. Регистрацию вольтамперограмм повторяли до тех пор, пока относительная разность высот пиков в двух последних вольтамперограммах не превышала 5 – 8 %.
На рис. 3.2. – 3.4 приведены вольтамперограммы анодного растворения тяжелых металлов.
| 0 0,2 0,4 0,6 0,8 1,0 Е, В
|
|
 Рис. 3.2. Анодная инверсионная вольтамперограмма образца речной воды, содержащей 5 мкг/л Сu2+, 0,5 мкг/л Cd2+, 15 мкг/л Pb2+и Zn2+ после электролитического концентрирования в течение 5 мин при 1,2 В. Рабочий электрод – СУ. Электрод сравнения – хлорсеребряный.

Рис. 3.3. Анодная инверсионная вольтамперограмма образца речной воды. 1 – фон, 2 – проба, 3 – проба с добавками градуировочных растворов. Время электролитического концентрирования – 5 мин при 1,2 В. Рабочий электрод – СУ. Электрод сравнения – хлорсеребряный.

Рис. 3.4. Анодная инверсионная вольтамперограмма образца речной воды, содержащей 5 мкг/л Сu2+ после электролитического концентрирования в течение 5 мин при 1,2 В. 1 – фон, 2 – проба, 3 – проба с добавками градуировочного раствора (1 мл). Рабочий электрод – СУ. Электрод сравнения – хлорсеребряный.
3.4 Обсуждение результатов
По результатам анализа с использованием метода добавок был построен калибровочный график, по которому были определены концентрации тяжелых металлов.
 Рис. 3.5. Калибровочный график (ионы меди)
Подобные расчеты были проведены и для других ионов (Pb, Zn) для различных проб.
В таблицах 3.2 и 3.3 приведены результаты анализа растворов сточных вод и водопроводной воды.
Таблица 3.1
Содержание тяжелых металлов в сточной воде
| № пробы сточных вод
|
Cср×10-2 мг/л
|
S×10-3
|
DCср×10-2 мг/л
|
m = (Cср±DCср)×10-2
|
| №1
|
| Zn
Pb
Cu
|
5,88
2,07
8,46
|
2,12
2,83
4,51
|
0,67
0,25
1,78
|
5,88± 0,67
2,07 ±0,25
8,46 ±1,78
|
| №2
|
| Zn
Pb
Cu
|
2,12
23,60
12,40
|
7,94
7,07
9,19
|
0,01
2,50
2,40
|
2,12± 0,01
23,60 ±2,50
12,40 ±2,40
|
| №3
|
| Zn
Cd
Pb
Cu
|
1,41
0,11
5,06
14,91
|
0,35
0,08
0,71
1,41
|
0,66
0,03
0,89
5,00
|
1,41±0,66
0,11±0,03
5,06±0,8914,9 ±5,00
|
Таблица 3.2
Содержание тяжелых металлов в водопроводной воде
| Тяжелые
металлы
|
Cср×10-2 мг/л
|
S×10-3
|
DCср×10-2 мг/л
|
m = (Cср ± DCср)×10-2
|
| №1
|
| Zn
Cd
Pb
Cu
|
1,32
0,25
0,96
6,81
|
3,18
0,18
0,14
0,75
|
0,41
0,02
0,07
0,33
|
1,32±0,41
0,25±0,02
0,96±0,07
6,81±0,33
|
| №2
|
| Zn
Cd
Pb
Cu
|
1,53
5,47×10-2
5,71
10,30
|
0,21
0,04
1,15
7,29
|
0,06
0,11×10-2
0,01
2,40
|
1,53±0,06
(5,47±0,11)×10-2
5,71±0,01
10,30±2,40
|
| № 3
|
| Zn
Cd
Pb
Cu
|
1,44
4,35×10-2
0,96
11,50
|
1,04
0,06
0,03
10,41
|
0,13
0,80×10-2
0,04
1,04
|
1,44±0,13
(4,35±0,80)×10-2
0,96±0,04
11,50±1,04
|
Следует отметить, что содержание тяжелых металлов в водопроводной воде в целом не превышает ПДК для воды.
Выводы
В результате проведенных исследований можно сделать следующие выводы:
1. Проведен анализ различных литературных данных по проблеме анализа микрограммовых количеств тяжелых металлов методом инверсионной вольтамперометрии;
2. С целью выработки методики исследования содержания тяжелых металлов проанализированы методики проведения анализа различных объектов окружающей среды методом инверсионной вольтамперометрии;
3. Разработана методика определения следовых количеств тяжелых металлов методом инверсионной вольтамперометрии исходя из имеющихся материалов и аппаратуры;
4. Проведено исследование различных проб водопроводной и сточных вод и проанализированы результаты, показывающие, что содержание тяжелых металлов (ионы меди, цинка и свинца) в водопроводной воде в целом соответствуют нормам ПДК.
Список литературы
1. Ф. Выдра, К. Штулик, Э. Юлакова. Инверсионная вольтамперометрия / пер. с чешского. В.А. Немова. // Под ред. Б. Я. Каплана. М.: Мир, 1980. с. 257 – 265.
2. Huderovd L., Stulik К., Talanta, 19, 1285 (1972).
3. Синякова С. И., Маркова И. В., Гальфаян Н. Г. Труды комис. по аналит. химии АН СССР, 1965, 15, с. 164
4. Zbinden С., Bull. Soc. Chim. Biol., 13, 35 (1931).
5. Rose J. W., de Mars R. D., Shain I., Anal. Chem., 28, 1768 (1956).
6. de Mars R. D., Shain I., Anal. Chem., 29, 1825 (1957).
7. Nikelly J. G., Cooke W. D., Anal. Chem., 29, 933 (1957).
8. Kalvoda R., Collect. Czech. Chem. Comm., 22, 1390 (1957).; Kalvoda R., Anal. Chim. Acta, 18, 132 (1958).
9. Barker G. C., Anal. Chim. Acta, 18, 118 (1958).; von Sturm F., Ressel M., Z. Anal. Chem., 186, 63 (1962).
10. Underkofler W. L., Shain I., Anal. Chem., 37, 218 (1965).
11. Lord S. S., O'Neill R. C., Rogers L. В., Anal. Chem., 24, 209 (1952).
12. Mamantov G., Papoff P., Delahay P., J. Am. Chem. Soc., 79, 4034 (1957).
13. Mamantov G., Papoff P., Delahay P., J. Am. Chem. Soc., 78, 2969 (1956).
14. Kopawica M., Vydra F., J. Electroanal. Chem., 31, 175 (1971).
15. Zieglerovd L., Stulik K., Dolezal J., Talanta, 18, 603 (1971).
16. Harrison P. R., Winchester J. W., Atmospheric Environment, 5, 863 (1971).
17. Neeb R., Saur D., Z. Anal. Chem., 222, 200 (1966).
18. Booth M. D., Brand M. J. D., Fleet В., Talanta, 17, 1059 (1970).
19. Kalvoda R., Chem. listy, 64, 3 (1970)1 66, 897 (1972).
20. Kalvoda R., Pouziti operacnich zesilovacu v chemicke instrumenta. SNTL, Praha, 1974.
21. Khasgiwale К. J., Parthasarathy R., Sankvar Das M., Anal. Chim. Acta, 59, 485 (1972)
22. Каплин А. А., Катюхин В. E., Стромберг А. Г. Завод. лаб., 1970, 36, с. 18.
23. Tyler J. F. C., Analyst, 89, 775 (1964).
24. Аверяскина Е.О., Ермаков С. С., Москвин Л. Н. Определение ртути в воздухе методом инверсионной вольтамперометрии c электрогенерированием йода / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
25. В. Н. Баталова, Э. А. Захарова, Г.Б. Слепченко, В.М. Пичугина, О.В. Рихерт. Аналитические проблемы определения ртути в пищевых продуктах методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
26. Стожко Н.Ю., Белышева Г.М., Инжеватова О.В., Камышов В.М. Определение марганца в винах методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
27. Брайнина Х.З, Стенина Л.Э, Малахова Н.А. Инверсионная вольтамперометрия в анализе объектов окружающей среды и пищевых продуктов / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004".
28. Булгакова О.Н., Халфина П. Д. Определение мышьяка в воде / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
29. Карбаинов Ю.А., Гиндуллина Т.М., Сутягина Г.Н. Определение меди, свинца, висмута в нефтях и нефтепродуктах методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
30. Носкова Г.Н., Иванова Е.Е., Чернов В.И., Толмачева Т.П. Одновременное определение йода, цинка, кадмия, свинца и меди в воде методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
31. Толмачева Т.П., Иванов И. Ю., Мержа А.Н. Определение ртути в почвах методом инверсионной вольтамперометрии / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
32. Заичко А.В., Носкова Г.Н., Иванова Е.Е., Толмачева Т.П. Определение мышьяка в пищевых продуктах методом инверсионной вольтамперометрии без применения инертного газа / Тезисы VII конференции "Аналитика Сибири и Дальнего Востока - 2004"
33. Д. Плембэк. Электрохимические методы анализа. - М.: Мир, 1985 - 496 с.
34. Виноградов А.П. Основные закономерности распределения микроэлементов между растениями и средой // Микроэлементы в жизни растений и животных. М.: Изд-во АН СССР, 1952.
35. Орлов Д.С. Микроэлементы в почвах и живых организмах // Соросовский Образовательный Журнал. 1998. № 1. С. 61.
36. Будников Г.К. Тяжелые металлы в экологическом мониторинге водных систем //Там же. № 5. С. 23.
37. Прохорова Г.В., Иванов В.М., Бондарь Д.А. Адсорбционная инверсионная вольтамперометрия: Анализ природных и биологических объектов // Вестн. МГУ. Сер. 2, Химия. 1998. Т. 39, № 4. С. 219–231.
38. Методика выполнения измерений массовой концентрации ионов кадмия, свинца, меди и цинка в питьевых, природных, морских и очищенных сточных водах методом инверсионной вольтамперометрии / «НПКФ АКВИЛОН»// www.water.ru
39. И.А. Глушко. Применение инверсионной вольтамперометрии в анализе кислотных вытяжек из почв для определения Cu, Pb и Cd. // Вестник удмуртского университета / Химия 2005 №8 с. 88 – 101.
40. Электроаналитические методы в контроле окружающей среды/ Р. Кальвода, Я. Зыка, К- Штулик и др. Пер. с англ. Под ред. Е. Я. Неймана. —М.: Химия, 1990. —240 с.
41. Полторанина Т.Н., Соколова О.Ю., Халфина П.Д. Инверсионно-вольтамперометрическое определение цинка, кадмия, свинца и меди при совместном присутствии в различных объектах окружающей среды
Приложения
Приготовление основных стандартных растворов металлов с концентрацией
1. Средства измерений, реактивы, оборудование
Весы лабораторные аналитические любого типа 2-го класса точности
Колбы мерные наливные : 2-1000-2 по ГОСТ 1770
Цилиндры мерные: 1-50 по ГОСТ 1770
Электроплитка
Стаканы термостойкие: В-1-150ТС по ГОСТ 25336
Вода бидистиллированная
Кислота азотная
Кадмий металлический КД-1 по ГОСТ 1467
Медь катодная по ГОСТ 859
Свинец металлический по ГОСТ 3778 или по ТУ 6-09-1490
Цинк металлический, гранулированный ЦВОО по ГОСТ 3640 или по ГОСТ 989
2. Приготовление растворов
2.1. Приготовление раствора азотной кислоты концентрации 0,1М
В мерную колбу вместимостью 1000 см3 вносят 100-200 см 3 бидистиллированной воды, цилиндром добавляют 8,3 см 3 концентрированной азотной кислоты, перемешивают, охлаждают и доводят до метки бидистиллированной водой.
2.2. Приготовление аттестованных растворов ионов кадмия, свинца, меди и цинка.
Основные аттестованные растворы металлов с концентрацией 1,0 мг/см3 готовят из чистых элементов (не менее 99,9%).
Навеску металла (медь, цинк, кадмий, свинец) 1,000 ± 0,001 г помешают в стакан вместимостью 150 см3, приливают 10 см3 конц. азотной кислоты, 10 см3 бидистиллированной воды и растворяют при нагревании.
К охлажденному раствору приливают 50 см3 0,1 М азотной кислоты (п. 3.3.23), раствор количественно переносят в мерную колбу вместимостью 1000 см3, доливают до метки тем же раствором азотной кислоты и перемешивают. Срок хранения растворов 1 год.
Основные аттестованные растворы металлов с концентрацией 1,0 мг/см3 предназначены для приготовления рабочих растворов с концентрациями. Аттестованные растворы с массовой концентрацией 100, 10 и 1 мкг/см3 готовят последовательным разбавлением основного аттестованного раствора.
Форма записи результатов анализа
| N пробы
|
Результат определения
|
Расхождение между параллельными определениями
|
Результат
анализа
|
| 1
|
| 2
|
| 3
|
| Среднее
|
|