Введение
Электрофильное замещение, несомненно, составляет самую важную группу реакций ароматических соединений. Вряд ли найдется какой-нибудь другой класс реакций, который так детально, глубоко и всесторонне исследован как с точки зрения механизма, так и с точки зрения применения в органическом синтезе. Именно в области электрофильного ароматического замещения впервые была поставлена проблема связи между структурой и реакционной способностью, которая является основным предметом изучения в физической органической химии. В общем виде этот тип реакций ароматических соединений может быть представлен следующим образом:
ArH + E+
 ArE + H+ ArE + H+

1. Литературный обзор
1.1 Электрофильное замещение в ароматическом ряду
Эти реакции характерны не только для самого бензола, но и вообще для бензельного кольца, где бы оно ни находилось, а также для других ароматических циклов — бензоидных и небензоидных. Реакции электрофильного замещения охватывают широкий круг реакций: нитрование, галогенирование, сульфирование и реакции Фриделя — Крафтса свойственны почти всем ароматическим соединениям; реакции типа нитрозирования и азосочетания присущи лишь системам с повышенной активностью; такие реакции, как десульфирование, изотопный обмен, и многочисленные реакции циклизации, которые с первого взгляда кажутся совсем различными, но которые также оказывается целесообразным отнести к реакциям того же типа.
1.1.1
Электрофильные агенты
Электрофильные агенты Е+
, хотя наличие заряда не обязательно, т.к. электрофил может быть и незаряженной электронодефицитной частицей (например, SO3
, Hg(OCOCH3
)2
и т.п.). Условно их можно разделить на три группы: сильные, средней силы и слабые.
Сильные электрофилы
NO2
+
(Ион нитрония, нитроил-катион); комплексы Cl2
или Br2
с различными кислотами Льюиса (FeCl3
, AlBr3
, AlCl3
, SbCl5
и т.д.); H2
OCl +
, H2
OBr +
, RSO2
+
, HSO3
+
, H2
S2
O7
. Сильные электропилы взаимодействуют с соединениями ряда бензола, содержащими как электронодонорные, так и практически любые электроноакцепторные заместители.
Электрофилы средней силы
Комплексы алкилгалогенидов или ацилгалогенидов с кислотами Льюиса (RCl.
AlCl3
, RBr.
GaBr3
, RCOCl.
AlCl3
и др.); комплексы спиртов с сильными кислотами Льюиса и Бренстеда (ROH.
BF3
, ROH.
H3
PO4
, ROH.
HF). Реагируют с бензолом и его производными, содержащими электронодонорные (активирующие) заместители или атомы галогенов (слабые дезактивирующие заместители), но обычно не реагируют с производными бензола, содержащими сильные дезактивирующие электроноакцепторные заместители (NO2
, SO3
H, COR,CN и др.).
Слабые электрофилы
Катионы диазония ArN+є
N, иминия CH2
=N+ H2
, нитрозония NO+
(нитрозоил-катион); оксид углерода (IY) СО2
(один из самых слабых электрофилов). слабые электрофилы взаимодействуют только с производными бензола, содержащими очень сильные электронодонорные заместители (+М)-типа (OH, OR, NH2
, NR2
, O- и др.).
1.1.2
Механизм электрофильного ароматического замещения
В настоящее время ароматическое электрофильное замещение рассматривается как двухстадийная реакция присоединения-отщепления с промежуточным образованием аренониевого иона, называемого σ-комплексом
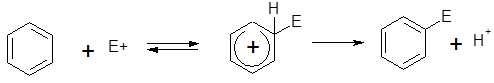
I-Аренониевый ион ( -комплекс), как правило, короткоживущий. Такой механизм получил название SE
Ar, т.е. SЕ
(аренониевый). В этом случае на первой стадии в результате атаки электрофила циклическая ароматическая 6-электронная π-система бензола исчезает и заменяется в интермедиате I на нециклическую 4-электронную сопряженную систему циклогексадиенильного катиона. На второй стадии вновь восстанавливается ароматическая -комплекс), как правило, короткоживущий. Такой механизм получил название SE
Ar, т.е. SЕ
(аренониевый). В этом случае на первой стадии в результате атаки электрофила циклическая ароматическая 6-электронная π-система бензола исчезает и заменяется в интермедиате I на нециклическую 4-электронную сопряженную систему циклогексадиенильного катиона. На второй стадии вновь восстанавливается ароматическая  -система за счет отщепления протона.Строение аренониевого иона I изображают различными способами: -система за счет отщепления протона.Строение аренониевого иона I изображают различными способами:
 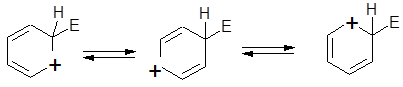
Наиболее часто употребляется первая формула. σ-комплекс будет гораздо лучше стабилизироваться донорными заместителями, находящимися в орто- и пара-положениях, чем донорными заместителями в мета-положении.
π
-Комплексы
Как известно, арены являются π-основаниями и могут образовывать донорно-акцепторные комплексы со многими электрофильными реагентами.Так, при пропускании сухих газообразных HCl или DCl в раствор бензола, толуола, ксилолов или других полиалкилбензолов в н-гептане при -78о
С удалось обнаружить образование молекулярных комплексов состава 1:1 (Г.Браун, 1952 г.).
Эти комплексы не окрашены, их растворы в ароматических углеводородах неэлектропроводны. При растворении газообразного DCl в бензоле, толуоле, ксилолах, мезитилене, пентаметилбензоле не происходит обмен H на D. Поскольку растворы комплексов не проводят электрический ток, они не являются ионными частицами, т.е. это не аренониевые ионы.
Такие донорно-акцепторные комплексы получили название π-комплексов. Например, кристаллы комплексов бензола с бромом или хлором состава 1:1 согласно рентгеноструктурным данным состоят из цепочек чередующихся молекул π-донора состава (C6
H6
) и акцептора (Cl2
,Br2
), в которых молекула галогена расположена перпендикулярно плоскости кольца вдоль оси, проходящей через его центр симметрии.
σ-комплексы (аренониевые ионы)
При введении в раствор HCl и DCl в алкилбензолах AlCl3
или AlBr3
раствор начинает проводить электрический ток. Такие растворы окрашены и их окраска изменяется от желтой до оранжево-красной при переходе от пара-ксилола к пентаметилбензолу. В системах ArH-DCl-AlCl3
или ArH-DF-BF3
атомы водорода ароматического кольца уже обмениваются на дейтерий. Электропроводность растворов определенно указывает на образование ионов в тройной системе арен-галогенводород-галогенид алюминия. Строение таких ионов было установлено с помощью ЯМР-спектроскопии на ядрах 1
Н и 13
С в системе ArH-HF(жидк)
-BF3
или ArH-HF-SbF5
в SO2
ClF при низкой температуре.
1.1.3
Классификация заместителей
Монозамещенные бензолы С6
Н5
Х могут быть более или менее реакционноспособны, чем сам бензол. Если в реакцию ввести эквивалентную смесь С6
Н5
Х и С6
Н6
, то замещение будет происходить селективно: в первом случае в реакцию будет вступать преимущественно С6
Н5
Х, а во втором случае - преимущественно бензол.
В настоящее время заместители делят на три группы с учетом их активирующего или дезактивирующего влияния, а также ориентации замещения в бензольном кольце.
1. Активирующие орто-пара-ориентирующие группы. К ним относятся: NH2
, NHR, NR2
, NHAc, OH, OR, OAc, Alk и др.
2. Дезактивирующие орто-пара-ориентирующие группы. Это галогены F, Cl, Br и I.
3. Дезактивирующие мета-ориентирующие группы. Эту группу составляют NO2
, NO, SO3
H, SO2
R, SOR, C(O)R, COOH, COOR, CN, NR3+
,CCl3
и др. Это ориентанты II-го рода.
Естественно, что существуют и группировки атомов промежуточного характера, обусловливающие смешанную ориентацию. Кним, например, относятся: CH2
NO, CH2
COCH3
, CH2
F, CHCl2
, CH2
NO2
, CH2
CH2
NO2
, CH2
CH2
NR3
+
, CH2
PR3
+
, CH2
SR2
+
идр.
1.2
Электрофильное замещение в π-избыточных гетероциклах
Фуран, пиррол и тиофен обладают значительной реакционной способностью по отношению к обычным электрофильным реагентам. В этом смысле они напоминают наиболее реакционно-способные производные бензола, такие, как фенолы и анилины. Повышенная чувствительность к электрофильному замещению вызвана несимметричным распределением заряда в этих гетероциклах, в результате чего на углеродных атомах цикла имеется больший отрицательный заряд, чем в бензоле. Фуран обладает несколько большей реакционной способностью, чем пиррол, а наименее реакционноспособен тиофен.
1.2.1
Электрофильное замещение пиррола
В то время как пиррол и его производные не склонны креакциям нуклеофильного присоединения и замещения, они очень чувствительны к электрофильным реагентам, и реакции пирролов с такими реагентами протекают практически исключительно как реакции замещения. Незамещенный пиррол, N- и С-моноалкилпирролы и в наименьшей степени С,С-диалкилпроизводные полимеризуются в сильнокислых средах, поэтому большинство электрофильных реагентов, использующихся в случае производных бензола, не применимы для пиррола и его алкилпроизводных.
Однако при наличии в пиррольном цикле электроноакцепторных групп, препятствующих полимеризации, например, таких, как сложноэфирная, становится возможным использование сильнокислых сред, нитрующих и сульфирующих агентов.
Протонирование
В растворе наблюдается обратимое присоединение протона по всем положениям пиррольного цикла. Наиболее быстро протонируется атом азота, присоединение протона по положению 2 проходит в два раза быстрее, чем по положению 3. В газовой фазе при использовании кислот умеренной силы, таких, как C4
H9
+
и NH4
+
, пиррол протонируется исключительно по атомам углерода, причем склонность к присоединению протона по положению 2 выше, чем по положению 3. Наиболее термодинамически стабильный катион - 2Н-пирролиевый ион - образуется при присоединении протона по положению 2 и определяемое значение рКа
для пиррола связано именно с этим катионом. Слабая N-основность пиррола обусловлена отсутствием возможности для мезомерной делокализации положительного заряда в 1H-пирролиевом катионе.
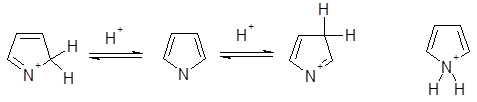
Значение рКа
определено для большого числа производных пиррола, а сам незамещенный пиррол — чрезвычайно слабое основание со значением рКа
-3,8. Основность пиррольного цикла весьма быстро увеличивается при введении алкильных заместителей, и для 2,3,4,5-тетраметилпиррола рКа
равен +3,7, что соответствует полному протонированию всех молекул пиррола в вышеприведенных условиях (для сравнения рКа анилина +4,6). Таким образом, алкильные группы оказывают необычайное стабилизирующее влияние на катионы - пирролы, содержащие трет-бутильные группы, при протонировании образуют стабильные кристаллические соли.
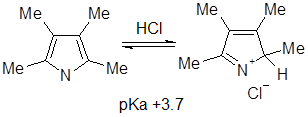 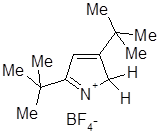
Реакции протонированных пирролов
2Н- и ЗН-Пирролиевые катионы в сущности представляют собой иминиевые ионы и, следовательно, обладают свойствами электрофилов. Эти катионы играют ключевую роль в процессах полимеризации и восстановления пирролов в присутствии кислот. При взаимодействии пирролов с гидрохлоридом гидроксиламина, сопровождающимся раскрытием цикла и образованием 1,4-диоксимов, вероятно, образуется более реакционноспособный ЗН-пирролиевый катион. Для защиты аминогруппы первичных аминов могут быть превращены в 1-К-2,5-диметилпирролы, затем защитная группа может быть удалена с помощью описанной выше реакции с гидроксиламином.

Нитрование
Нитрующую смесь, применяемую для нитрования производных бензола, нельзя использовать в случае пиррола, поскольку это приводит к его полному разложению. Однако нитрование пиррола возможно при использовании ацетилнитрата при низких температурах, причем преимущественно образуется 2-нитро-пиррол. Ацетилнитрат получают при смешивании дымящей азотной кислоты с уксусным ангидридом, и в результате образуется уксусная кислота и достигается удаление сильной минеральной кислоты. При нитровании пиррола с использованием ацетилнитрата активность положения 2 в 1,3 • 105
, а положения 3 в 3 • 104
раза выше активности бензола.

Введение заместителя к атому азота увеличивает долю продукта нитрования по положению 3 в смеси продуктов реакции: так, введение метального заместителя обусловливает получение смеси продуктов β- и α-нитрования в соотношении 1:3. Более объемная трет-бутильная группа приводит даже к обращению относительной реакционной способности — продукты β- и α-нитрования образуются в соотношении 4:1. Полного подавления реакции нитрования по α-положению пиррола можно достигнуть при введение к атому азота триизопропилсилильной (TIPS) группы; использование последней чрезвычайно важно при синтезе 3-производных, так как в последствии она может быть легко удалена.
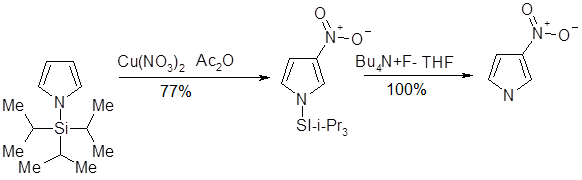
Сульфирование и реакции с использованием других серосодержащих электрофильных реагентов
Для сульфирования пирролов используются мягкие сульфирующие агенты, не обладающие свойствами кислоты; так, комплекс пиридин — триоксид серы мягко превращает пиррол в пиррол-2-сульфонат .

Реакции с использованием других серосодержащих электрофильных реагентов, например сульфинилирование пиррола, тиоцианирование пиррола и 1-фенилсульфоиилпиррола, позволяют получать серосодержащие производные пиррола с атомом серы в более низкой степени окисления.
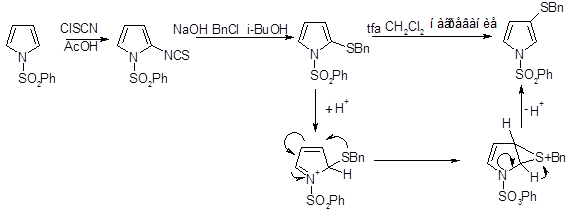
Катализируемая кислотой перегруппировка производных пиррола, содержащих серосодержащий заместитель в α-положении (продукт кинетически контролируемого электрофильного замещения), позволяет получать соответствующие β-изомеры.
Галогенирование
Галогенирование пиррола протекает настолько легко, что, если специальным образом не контролировать течение реакции, образуются исключительно стабильные тетрагалогенопроизводные. Попытки провести моногалогенирование простых алкилпирролов оказались безуспешными, поскольку при этом образуются чрезвычайно реакционноспособные пиррилалкилгалогениды - продукты галогенирования боковой цепи.
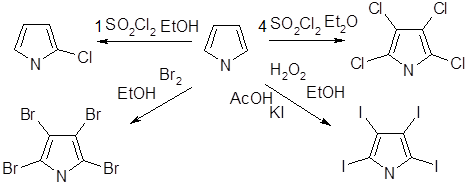
2-Бром - и 2-хлорпирролы — нестабильные соединения, которые можно получить прямым галогенированием пиррола. Использование 1,3-дибром-5,5-диметилгидантоина в качестве бромирующего агента приводит к образованию 2-бром - и 2,5-дибромпирролов; продукты бромирования стабилизируютнемедленным превращением в N-трет-бутилоксикарбонильные производные. Бромирование N-трет-бутилоксикарбонилпиррола с использованием N-бромсукцинимида приводит к 2,5-дибромпроизводному
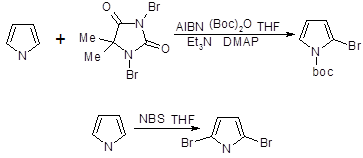
При монобромировании и моноиодировании N-триизопропилсилилпиррола образуются практически исключительно 3-галогенозамещенные пирролы, а использование двух эквивалентов N-бромсукцинимида позволяет получить 3,4-дибромпроизводное
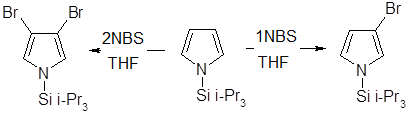
Ацилирование
Прямое ацетилирование пиррола уксусным ангидридом при 200 °С приводит к образованию 2-ацетилпиррола с примесью небольшого количества 3-ацетилпирола; N-ацетилпиррол в этих условиях вовсе не образуется N-Ацетилпиррол можно получить с высоким выходом при нагревании пиррола с N-ацетилимидазолом.
Алкильные заместители облегчают процесс ацилирования по атому углерода: так, 2,3,4-триметилпиррол превращается в 5-ацетилпроизводное даже при кипячении в уксусной кислоте. Более реакционноспособные трифторуксусный ангидрид и трихлорацетилхлорид реагируют с пирролом даже при комнатной температуре с образованием продуктов 2-ацилирования, которые в результате гидролиза или алкоголиза обеспечивают удобный синтетический подход к пиррол-2-карбоновым кислотам или их эфирам.
Сильные электроноакцепторные заместители (мета-ориентирующие группы) в α -положении пиррольного кольца склонны изменять присущую пирролу региоселективность в реакциях электрофильного замещения — последующее замещение протекает по положению 4, а не по свободному α-положению.
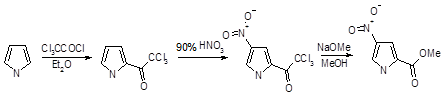
Алкилирование
Моноалкилпроизводные пиррола (по атому углерода) не удается получить прямой реакцией пиррола с алкилгалогенидами как в результате катализируемого кислотой Льюиса алкилирования, так и в отсутствие катализатора. Пиррол не вступает в реакцию с метилиодидом при температурах ниже 100˚С, однако при температурах выше 150˚С в результате серии превращений образуется сложная смесь, состоящая главным образом из продуктов полимеризации и некоторого количества полиметилированных производных пиррола. Более реакционноспособный аллилбромид реагирует с пирролом при комнатной температуре, однако в результате этого взаимодействия образуется смесь различных аллилпирролов (от моно - до тетразамещенных) одновременно с продуктами олигомеризации и полимеризации. Наиболее гладко пиррол моноалкилируется сопряженными енонами, содержащими уходящую группу в β-положении.
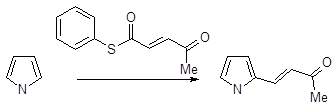
1.2.2
Электрофильное замещение тиофена
Как и следовало ожидать, для тиофена характерны реакции, свойственные ароматическим соединениям типа бензола. Тиофен галогенируется, нитруется, сульфируется и ацилируется аналогично фурану и пирролу, но в более жестких условиях.
Протонирование. Реакции протонированных тиофенов
Вследствие высокой стабильности тиофенов в их реакциях электрофильного замещения могут быть успешно использованы комбинации ряда реагентов с сильными кислотами, обычно приводящие к кислотно-катализируемому разложению и полимеризации фуранов и пирролов. Изучение катализируемого кислотами обмена на атом водорода других групп, например кремния или ртути , показало, что скорость атаки по положению 2 примерно в 1000 раз выше, чем по положению 3. Так, для 2,5-ди-трет-бутилтиофена рКа
при образовании соли за счет протонирования по положению 2 составляет —10,2.При обработке тиофена горячей фосфорной кислотой получается тример, строение которого предполагает, что на первой стадии образование связи С—С
вдет через α-протонированный катион.

Нитрование
Нитрование тиофена следует проводить в отсутствие азотистой кислоты, которая может привести к взрыву; наиболее удобно использование ацетилнитрата или тетрафторбората нитрония. Наряду с преимущественным образованием 2-нитросоединения получают и 3-изомер с выходом -10%. Дальнейшее нитрование как 2-, так и 3-нитротио-фенов также приводит к образованию смесей: из 2-нитротиофена образуются эквивалентные количества 2,4- и 2,5-динитротиофенов, а из 3-изомера — в основном 2,4-динитротиофен. Аналогично, предсказуемая смесь изомеров образуется при нитровании замещенных тиофенов: например, 2-метилтиофен дает смесь 2-метил-5- и 2-метил-З-нитротиофенов, а 3-метилтиофен -4-метил-2- нитро- и 3-метил-2-нитротиофены в соотношении 4:1 в каждом случае.
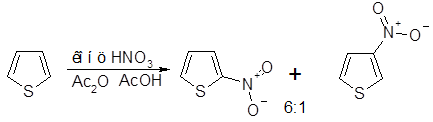
Сульфирование
Получение тиофен-2-сульфокислоты при сульфировании серной кислотой уже давно известно, однако использование комплекса пиридинсульфотриоксид, возможно, более удобный метод. 2-Хлорсульфонилирование и 2- тиоцианирование также достаточно эффективны.
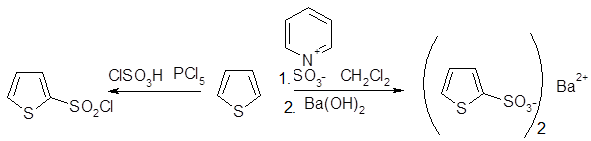
Галогенирование
Галогенирование тиофена происходит очень быстро и как при комнатной температуре, так и при -30 °С в темноте легко проходит тетразамещение . Скорость галогенирования тиофена при 25 °С примерно в 108
раз больше, чем бензола. Образование как 2,5-дибром - и 2,5-дихлортиофенов, так и 2-бром- и 2-иодтиофенов гладко происходит в различных контролируемых условиях. Контролируемое бромирование 3-бромтиофена дает 2,3-ди-бромтиофен.

Трибромирование тиофена гладко протекает в 48%-ном растворе НВг. Давно известно, что при обработке полигалогенотиофенов цинком и кислотой удается селективно удалять α-галоген, что делает доступным получение 3-бромтиофена, а 3,4-дибромтиофен аналогично образуется при восстановлении тетрабромида. При использовании боргидрида натрия из 2,3,5-трибромтиофена получают 2,3-дибромтиофен (в присутствии катализатора Pd(0))или 2,4-дибромтиофен (без катализатора).

Моноиодирование α-замещеиных тиофенов, независимо оттого, оказывают ли эти заместители активирующее или дезактивирующее влияние, идет по второму α-положению при действии иода и иодбензолдиацетата. 3-Алкилтио-фены можно монобромировать или моноиодировать по положению 2 при использовании N-бромсукцинимида или иода в присутствии оксида ртути(II) соответственно.
Ацилирование
Ацилирование по Фриделю-Крафтсу наиболее часто используется для тиофенов и обычно дает хорошие выходы в контролируемых условиях. Несмотря на то, что при взаимодействии тиофена с AlCl3
получаются смолы, их образования можно избежать, если добавлять катализатор к тиофену и ацилирующему агенту. Наиболее часто в качестве катализатора используют хлорид олова. Эффективный метод — ацилирование ангидридами, катализируемое фосфорной кислотой. В реакциях с ацетил-п-толуолсульфонатами в отсутствие катализаторов с высоким выходом образуется 2-ацетилтиофен. При формилировании тиофена по Вильсмейеру получают 2-формилтиофен. 2-Формилпроизводное образуется также из 3-фенилтиофена в условиях реакции Вильсмейера.

Алкилирование
Алкилирование проходит достаточно легко, однако редко используется в препаративной химии. Одним из примеров может служить эффективное 2,5-бис-трет-бутилирование тиофена.
Конденсация с альдегидами и кетонами
Кислотно-катализируемые реакции тиофена с альдегидами и кетонами неприменимы для получения гидроксиалкилтиофенов, так как они нестабильны в условиях реакции. Однако можно осуществить хлоралкилирование, а при использовании хлорида цинка даже тиофены, имеющие электроноакцепторные заместители, вступают в эту реакцию. Необходимо тщательно подбирать условия, так как возможно образование как ди-2-тиенилметанов, так и 2,5-бис(хлорметил)тиофена.
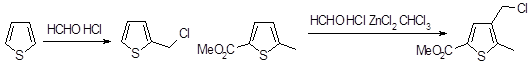
1.2.3 Электрофильное замещение фурана
Среди трех пятичленных систем с одним гетероатомом, фуран представляет собой «наименее ароматическое» соединение и, как таковой, проявляет наибольшую склонность к реакциям присоединения.
Это верно в отношении как взаимодействия с обычными электрофильными реагентами, используемыми в реакциях замещения, так и процессов типа реакции Дильса-Альдера.
Нитрование
Чувствительность фуранов не позволяет использовать концентрированную кислотную нитрующую смесь. При взаимодействии фурана или замещенных фуранов с ацетилнитратом первоначально образуются неароматические аддукты, в которых образованию продуктов замещения препятствует процесс нуклеофильного присоединения ацетата к катионному интермедиату, обычно по положению 5. Ароматизация с потерей уксусной кислоты, приводящая к образованию нитропроизводного, происходит под действием растворителя; наилучшие результаты достигаются при использовании слабого основания, такого, как пиридин.
Дальнейшее нитрование 2-нитрофурана дает 2,5-ди-нитрофуран в качестве основного продукта реакции.

Сульфирование
Фуран и алкилфураны разлагаются под действием обычных сильных кислот, однако можно использовать комплекс пиридинсульфотриоксид, под действием которого происходит дизамещение фурана даже при комнатной температуре.

Галогенирование
Фуран энергично реагирует с хлором и бромом при комнатной температуре с образованием полигалогенированных соединений, но не реагирует с иодом. В более контролируемых условиях можно получить 2-бромфуран. реакция, по-видимому, протекает через образование 1,4-дибром-1,4-дигидроаддукта, поскольку такие частицы были действительно обнаружены при низкой температуре с использованием спектроскопии ПМР. В реакции с бромом в диметилформамиде при комнатной температуре гладко образуются 2-бром - и 2,5-дибромфураны.
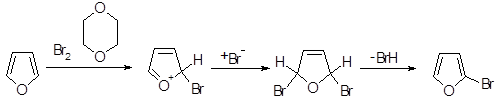
Ацилирование
Для ацилирования фуранов по Фриделю - Крафтсу ангидридами или галогенангидридами карбоновых кислот обычно необходимо присутствие кислоты Льюиса (часто трифторида бора), хотя реакция с ангидридом трифторуксусной кислоты не требует катализатора. Было показано, что при ацилировании фуранов в условиях катализа хлоридом алюминия α-положение проявляет реакционную способность, в 7 • 104
раз большую, чем реакционная способность β-положения. 3-Алкилфураны замещаются главным образом по положению 2; 2,5-диалкил фураны могут быть проацилированы по β-положению, хотя обычно большим трудом.
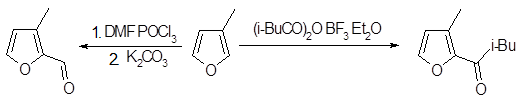
Формилирование фуранов по Вильсмейеру обеспечивает удобный подход к формилфуранам, хотя не меньшую роль играют легкая доступность фурфурола в качестве исходного материала, а также важны методы, включающие литиирование фуранов. Формилирование замещенных фуранов происходит согласно правилу предпочтительного образования α-замещенных производных, несмотря на все другие факторы; так, и 2-метилфуран, и метиловый эфир фуран-3-карбоновой кислоты дают 5-альдегид, а 3-метилфуран превращается главным образом в 2-альдегид.
2. Практическая часть
2.1 Пиримидин
Как известно, гетероароматические системы весьма отчетливо подразделяются на π-избыточные и π-дефицитные. Первым свойственны реакции электрофильного замещения, окисления, тогда как вторые реагируют главным образом с нуклеофилами, трудно окисляются, но сравнительно легко восстанавливаются. гетероароматическая система, обладающая одновременно свойствами π-избыточных и π-дефицитных соединений является перимидин, химическая амфотерность которого делает его интереснейшим объектом исследования.
2.1.1 Реакции электрофильного замещения
Перимидин является одним из наиболее активных по отношению к электрофильным агентам гетероциклов, что объясняется его высокой π-донорной способностью и большим отрицательным π-зарядом в орто-и пара-положениях нафталинового кольца. Именно по ним и протекают все реакции электрофильного замещения; до сих пор не известно случаев атаки электрофилами положений 5 и 8. Реакции электрофильного замещения в перимидинах очень чувствительны к стерическим помехам со стороны N-заместителя. Лишь небольшие по размерам частицы (D+
, с большим трудом Сl+
) могут вступать в положения 4 и 9 при наличии соседних N-метильных групп.
Ацилирование
Перимидин - единственная гетероароматическая система с пиридиновым атомом азота, подвергающаяся сравнительно легкому ацилированию по Фриделю — Крафтсу. Ацилирование лучше всего проводить с помощью карбоновых кислот в среде полифосфорной кислоты (ПФК). Для соединений с незамещенной группой NH реакция имеет ярко выраженный кинетический и термодинамический контроль. При 70—85° образуется главным образом 6(7)-ацилпроизводное (55—85%) наряду с небольшим количеством 9-изомера. При 120—150° единственным продуктом реакции становятся 4(9)-ацилперимидины. Одной из причин повышенной устойчивости последних является наличие в них прочной внутримолекулярной водородной связи.
Нитрование
В зависимости от количества и концентрации азотной кислоты перимидины нитруются (лучше всего в среде уксусной кислоты) до моно-, ди-, три- и тетранитропроизводиых, а ацеперимидины — до моно-и динитропроизводиых. Первое нитрование перимидинов со свободной группой NH сопровождается осмолением, что снижает выход. Так, перимидин нитруется действием 1 моля HNO3
, образуя 4(9)- и 6(7)-нитропроизводные в соотношении 2,5 : 1 при общем выходе 30%.
Галогенирование
Хлорирование перимидинов, сульфурилхлоридом в уксусной кислоте и N-хлорбензотриазолом (ХБТ) в
апротонной среде. Хлорирование перимидина действием моля SO2
Сl2
, приводит к образованию 6(7)- и 4(9)-хлорзамещенных в соотношении 8:1. При действии 2 молей SO2
Сl2
образуется сложная смесь моно-, ди-и трихлорперимидинов, а 3 молей SO2
Сl2
- 4,6,7-трихлорперимидии с высоким выходом. Получить с помощью SO2
Сl2
тетрахлорперимидин не удалось, но 2-метилперимидин хлорируется избытком SO2
Сl2
дотетра-хлорпроизводного.
2.2 Синтез 7(6)ацетил перимидина исходя из 1, 8-Диаминонафталина

1, 8-Диаминонафталин (7,9 г, 0,05 моль) кипятят 1 ч с 15 мл муравьиной кислоты. Смесь разбавляют вдвое водой, кипятят 2—3 раза с активированным углем, фильтрат охлаждают и нейтрализуют 25%-ным раствором аммиака. Выпавший осадок отфильтровывают, хорошо промывают холодной водой и высушивают на воздухе, размазывая его тонким слоем на поверхности стеклянной пластинки. Ввиду мелкокристалличности перимидина даже хорошо отжатый на фильтре продукт содержит значительное количество воды. При высушивании в сушильном шкафу сырой продукт окисляется. Выход близок к количественному. Перимидин представляет собой желто-зеленые кристаллы с Тпл 224-225’С
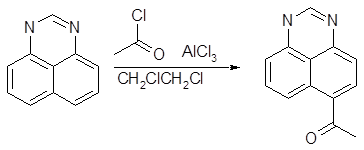
В коническую колбу емкостью 150мл, снабженную магнитной мешалкой и хлоркальциевой трубкой помещают 7,9г безводного хлористого алюминия, 0,52мл хлористого ацетила в 20мл дихлорэтана и в течение 1часа при перемешивании добавляют 1г перимидина, поддерживая при этом температуру 18-20’С. После добавления последней порции перимидина перемешивание продолжают еще 2часа (контроль ТСХ, проявляют хроматограмму парами иода). Затем реакционную массу тонкой струей выливают в воду (осторожно вспенивание и разогревание). Чтобы избавится от избытка дихлорэтана смесь нагревают. Выпавший кристаллический осадок фильтруют и промывают водой.
Экспериментальные данные приведены в таблицах
| Названия веществ и формула |
Характеристика исходных веществ |
Количества исходных веществ |
| М масса |
Физиол.
действие
|
Константы |
Кислоты и щелочи |
По методике |
По
Уравн.
реакции
|
Избыток
В
молях
|
| Тпл |
Ткип |
D4
20
N20
4
|
% конц |
D20
|
Грамм
Данной
конц
|
100% |
В молях |
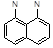
1, 8-Диаминонафталин
|
158 |
Канцероген |
117 |
- |
- |
7,9 |
7,9 |
0,05 |
0,05 |
1 |

Муравьиная кислота
|
46 |
Обладает отвлекающим и раздражительным действ. |
8,4 |
100,7 |
1,220
1,3714
|
18,3 |
18,3 |
0,4 |
0,05 |
8 |
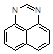
перимидин
|
168 |
канцероген |
224 |
- |
- |
1 |
1 |
0,006 |
0,006 |
1 |
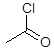
Ацетил хлористый
|
78 |
Токсическое
|
- |
182 |
1,1175
1,4627
|
0,5148 |
0,5148 |
0,0066 |
0,006 |
1,1 |
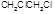 |
98 |
Токсическое |
-35,36 |
83,47 |
1,2351
1,4448
|
- |
- |
- |
- |
- |
AlCl3
Алюминий хлористый безводный
|
133,5 |
Токсическое |
172 |
179 |
- |
7,9 |
7,9 |
0,054 |
- |
9 |
| Название вещества и формула |
Константы |
Выход |
| Тпл |
Ткип |
n20
D
|
Масса, г |
% от теор |
% от указанного в методике |
Теорет-
ический
|
перимидин
|
224 |
Разлагается |
- |
8,316 |
99% |
99% |
99% |
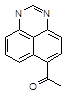 |
176 |
Разлагается |
- |
0,756 |
60% |
92% |
65% |
В процессе выполнения работы выполнялись следующие задачи:
1. Изучение и систематизация сведений об ароматическом электрофильном замещении
2. Изучение особенностей электрофильного замещения π-избыточных гетероциклов на примере фурана тиофена и пиррола
3. Изучение физических и химических свойств перимидина Синтез 7(6)ацетил перимидина исходя из 1, 8-Диаминонафталина
Заключение
Известно, что пиримидин относится к так называемым π-амфотерным системам, т.е. обладает одновременно свойствами ярко π-избыточных и π-дефицитных соединений. Поэтому, пиримидин и его производные способны вступать как в реакции нуклеофильного, так и в реакции электрофильного замещения. C другой стороны, имеются данные о биологической активности различных производных перимидина. Некоторые производные являются депрессантами и эффективными стимуляторами центральной нервной системы. 2-Аминоперимидины обладают противомикробнойактивностью, а 2-ациламиноперимидины – фунгицидным действием. Таким образом, продолжение изучения реакционной способности пиримидина и синтез новых функциональных производных этого гетероцикла является весьма перспективным и полезным направлением. Практическая часть курсовой работы состояла в получении 7(6)ацетил пиримидина, являющемся ценным реагентом для органического синтеза. На первой стадии синтеза для получения чистого пиримидина важную роль играет предварительная перегонка 1-8нафталиндиамина, так как перимидин плохо поддается перекристаллизации. Был получен пиримидин, пригодный для дальнейшего использования. Выход составил 99% от теоретического. Вторая стадия - ацилирование по Фриделю-Крафтсу важно не допустить попадания в реакционную смесь даже следов воды, для этого применялась хлоркальциевая трубка. Выход ацетил пиримидина составил 60% или 0,756г . потери связанны с частичной растворимостью соединения в воде.
Список литературы
1. Пожарский А. Ф., Анисимова В. А., Цупак Е. Б. Практические работы по химии гетероциклов // Изд-во РГУ. - 1988. - 158 с.
2. Дальниковская В. В., Комиссаров И. В., Пожарский А. Ф., Филиппов И. Т. Перимидины // Хим.-фарм. журнал, 1978, № 7, С. 85.
3. Успехи химии 1981 выпуск 9 с.1559-1594
4. Л. Пакетт Основы современной химии гетероциклических соединений Изд-во мир – 1968 с.97-134.
5. А.Е.Агрономов Ю.С. Шабаров Лабораторные работы в органическом практикуме М. Химия 1974.
6. В.И. Ивановский Химия гетероциклических соединений М Высшая школа 1978.
7. А.А. Потехина Свойства органических соединений Л. Химия 1984.
8. Дж. Джоуль К. Милс Химия гетероциклических соединений М Мир 2004.
9. И.И. Грандберг Органическая химия Дрофа 2002 А.Н.Несмеянов Н.А. Несмеянов Начала органической химии М 1969.
|