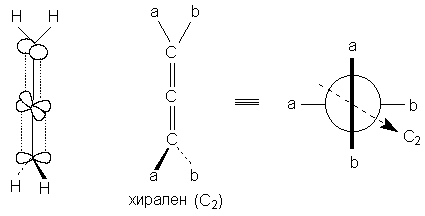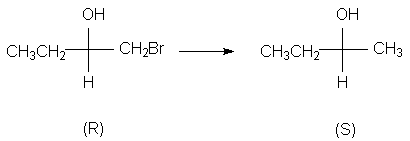Курсовая работа: Оптическая изомерия
|
Название: Оптическая изомерия Раздел: Рефераты по химии Тип: курсовая работа |
КУРСОВАЯ РАБОТА Тема: "Оптическая изомерия" Оглавление Введение 1. Оптическая активность 1.1 Оптически активные вещества 1.2 Физические причины оптической активности 1.2 а. Феноменологическая модель 1.2 б. Квантовая теория 1.2 в. Корпускулярная теория 2. Хиральные молекулы 2.1 Точечные группы симметрии 2.1 а. Собственная ось симметрии 2.1 б. Несобственная ось симметрии 2.1 в. Типы точечных групп симметрии 2.2 Симметричное определение хиральности 2.3 Типы хиральности 3. Номенклатура энантиомеров 3.1 По конфигурации: R - и S 3.2 По оптической активности: +/- 3.3 По конфигурации: в - и L- 4. Методы определения конфигурации 4.1 Определение абсолютной конфигурации 4.1 а. Дифракция рентгеновских лучей 4.1 б. Теоретический расчет оптического вращения 4.2 Определение относительной конфигурации 4.2 а. Химическая корреляция 4.2 б. Установление относительной конфигурации с помощью физических методов 5. Методы разделения энантиомеров 5.1 Расщепление через диастереомеры 5.2 Хроматографическое расщепление 5.3 Механическое расщепление 5.4 Ферментативное расщепление 5.5 Установление оптической чистоты Заключение Литература ВведениеСреди органических соединений встречаются вещества, способные вращать плоскость поляризации света. Это явление называют оптической активностью, а соответствующие вещества - оптически активными . Оптически активные вещества встречаются в виде пар оптических антиподов - изомеров, физические и химические свойства которых в обычных условиях одинаковы, за исключением одного - знака вращения плоскости поляризации. (Если один из оптических антиподов имеет, например, удельное вращение (+20 о , то другой - удельное вращение - 20 о ). Оптическая изомерия появляется тогда, когда в молекуле присутствует асимметрический атом углерода; так называют атом углерода, связанный с четырьмя различными заместителями. Возможны два тетраэдрических расположения заместителей вокруг асимметрического атома. Обе пространственные формы нельзя совместить никаким вращением; одна из них является зеркальным изображением другой:
Так же этот вид изомерии называют оптической изомерией , зеркальной изомерией или энантиомерией . Обе зеркальные формы составляют пару оптических антиподов или энантиомеров . В 1815 французский физик Жан Батист Био и немецкий физик Томас Зеебек установили, что некоторые органические вещества (например, сахар или скипидар) обладают свойством вращать плоскость поляризации света, в кристаллическом, в жидком, растворенном и даже газообразном состоянии (Впервые это явление обнаружил в 1811г. французский физик Франсуа Доминик Араго у кристаллов кварца). Так было доказано, что оптическая активность может быть связана не только с асимметрией кристаллов, но и с каким-то неизвестным свойством самих молекул. Оказалось, что некоторые химические соединения могут существовать в виде как право-, так и левовращающих разновидностей, причем самый тщательный химический анализ не обнаруживает между ними никаких различий. Это был новый тип изомерии, которую назвали оптической изомерией. Оказалось, что кроме право - и левовращающих, есть и третий тип изомеров - оптически неактивные. Это обнаружил в 1830 знаменитый немецкий химик Йёнс Якоб Берцелиус на примере виноградной (дигидроксиянтарной) кислоты НООС-СН (ОН) - СН (ОН) - СООН: эта кислота оптически неактивна, а винная кислота точно такого же состава обладает в растворе правым вращением. Позднее была открыта и не встречающаяся в природе "левая" винная кислота - антипод правовращающей. Различить оптические изомеры можно с помощью поляриметра - прибора, измеряющего угол поворота плоскости поляризации. Для растворов этот угол линейно зависит от толщины слоя и концентрации оптически активного вещества (закон Био). Для разных веществ оптическая активность может изменяться в очень широких пределах. Так, в случае водных растворов разных аминокислот при 25° С удельная активность (она обозначается как в и измеряется для света с длиной волны 589 нм при концентрации 1 г/мл и толщине слоя 10 см) равна - 232° для цистина, - 86,2° для пролина, - 11,0° для лейцина, +1,8° для аланина, +13,5° для лизина и +33,2° для аспарагина. Современные поляриметры позволяют измерять оптическое вращение с очень высокой точностью (до 0,001°). Подобные измерения позволяют быстро и точно определить концентрацию оптически активных веществ, например, содержание сахара в растворах на всех стадиях его производства - начиная от сырых продуктов и кончая концентрированным раствором и патокой. Оптическую активность кристаллов физики связывали с их асимметричностью; полностью симметричные кристаллы, например, кубические кристаллы поваренной соли оптически неактивны. Причина же оптической активности молекул долгое время оставалась совершенно загадочной. Первое открытие, проливавшее свет на это явление, сделал в 1848 никому тогда не известный Луи Пастер. Пастер, который выделил два антипода винной кислоты, которые получили название энантиомеров (от греч. enantios - противоположный). Пастер ввел для них обозначения L - и D-изомеров (от латинских слов laevus - левый и dexter - правый). Позднее немецкий химик Эмиль Фишер связал эти обозначения со строением двух энантиомеров одного из наиболее простых оптически активных веществ - глицеринового альдегида ОНСН2-СН (ОН) - СНО. В 1956 по предложению английских химиков Роберта Кана и Кристофера Ингольда и швейцарского химика Владимира Прелога для оптических изомеров были введены обозначения S (от лат. sinister - левый) и R (лат. rectus - правый); рацемат обозначают символом RS. Однако по традиции широко используются и старые обозначения (например, для углеводов, аминокислот). Следует отметить, что эти буквы указывают лишь на строение молекулы ("правое" или "левое" расположение определенных химических групп) и не связаны с направлением оптического вращения; последнее обозначают знаками плюс и минус, например, в (-) - фруктоза, в (+) - глюкоза. Теория, объясняющая отличие друг от друга молекул антиподов была создана голландским ученым Вант-Гоффом. Согласно этой теории, молекулы, как и кристаллы, могут быть "правыми" и "левыми", являясь зеркальным отражением друг друга. Подобные структуры, которые отличаются друг от друга как правая рука от левой, получили название хиральных (от греч. heir - рука). Таким образом, оптическая активность - следствие пространственной изомерии (стереоизомерии) молекул. оптическая изомерия эвантиомер хиральность Теория Вант-Гоффа, заложившая основы современной стереохимии, завоевала общее признание, а ее создатель в 1901 стал первым лауреатом Нобелевской премии по химии. 1. Оптическая активностьОптическая активность - это способность среды (кристаллов, растворов, паров вещества) вызывать вращение плоскости поляризации проходящего через нее оптического излучения (света). Впервые оптическая активность была обнаружена в 1811 г.Д. Араго в кристаллах кварца. В 1815 г.Ж. Бои открыл оптическую активность чистых жидкостей (скипидара), а затем растворов и паров многих, главным образом органических веществ. Ж.Био установил, что поворот плоскости поляризации происходит либо по часовой стрелке, либо против нее, если посмотреть навстречу ходу лучей света и в соответствии с этим разделил оптически активные вещества на правовращающие (вращающие положительно, т.е. по часовой стрелке) и левовращающие (отрицательно вращающие) разновидности. Наблюдаемое значение угла поворота плоскости поляризации в случае раствора связано с толщиной образца и концентрацией оптически активного вещества. Оптически активными веществами называют лишь те вещества, которые проявляют естественную оптическую активность. Существует также и искусственная или наведенная оптическая активность. Ее проявляют оптически неактивные вещества при помещении в магнитное поле (эффект Фарадея). 1.1 Оптически активные веществаОптически активные вещества подразделяются на два типа. К первому типу относятся вещества, которые оптически активны лишь в кристаллической фазе (кварц, киноварь). Ко второму типу относятся вещества, которые оптически активны в любом агрегатном состоянии (например, сахара, камфара, винная кислота). У соединений первого типа оптическая активность является свойством кристалла как целого, но сами молекулы или ионы, составляющие кристалл, оптически неактивны. Кристаллы оптически активных веществ всегда существуют в двух формах - правой и левой; при этом решетка правого кристалла зеркально симметрична решетке левого кристалла и никакими поворотами и перемещениями левый и правый кристаллы не могут быть совмещены друг с другом. Оптическая активность правой и левой форм кристаллов имеет разные знаки и одинакова по абсолютной величине (при одинаковых внешних условиях). Правую и левую форму кристаллов называют оптическими антиподами. У соединений второго типа оптическая активность обусловлена дисимметрическим строением самих молекул. Если зеркальное отображение молекулы никакими вращениями и перемещениями не может быть наложено на оригинал, молекула оптически активна; если такое наложение осуществить удается, то молекула оптически неактивна. (Под зеркалом понимают отражатель, лежащий вне молекулы, и отражение дает отображение всей молекулы). Асимметрические молекулы и дисcимметрические молекулы не одно и то же. Асимметрические молекулы не имеют никаких элементов симметрии, тогда как в дисcимметрических молекулах некоторые элементы симметрии остаются. Диcсимметрия есть нарушение максимальной симметрии объекта. Оптическую активность проявляют все асимметрические молекулы, но далеко не все диссимметрические молекулы. Оптическая активность связана лишь с дисcимметрией, обусловливающей несовместимость объекта с его зеркальным отображением. Такой вид диссимметрии, получил название хиральность. Хиральные объекты несовместимы в пространстве и представляются как зеркальные отображения друг друга. Оптически активная молекула хиральна, а оптически неактивная ахиральна, однако если молекулу нельзя совместить с ее зеркальным отображением, то зеркальное отображение соответствует другой, отличной молекуле, которую, в принципе, можно синтезировать. Синтезированное зеркальное отображение хиральной молекулы будет ее реальным оптическим изомером. Чистое оптически активное соединение имеет только два оптических изомера (т.к. каждому объекту соответствует лишь одно зеркальное отображение). Оптические изомеры называются энантиомерами (или иногда энантиоморфами). Удельное вращение энантиомеров одинаково по абсолютной величине и противоположно по знаку: один энантиомер левовращающий, а второй правовращающий. Кроме знака вращения все другие физические и химические свойства энантиомеров в газовой фазе, а также в ахиральных жидких средах одинаковы. Однако, если жидкая среда хиральна (например, в раствор добавлен хиральный реагент или катализатор, или сам растворитель хирален) свойства энантиомеров начинают различаться. При взаимодействии с другими хиральными соединениями, отзывающимися на зеркальную изомерию молекул, энантиомеры реагируют с различными скоростями. Особенно ощутимо различие в физиологическом и биохимическом действии энантиомеров, что связано с энантиомерией биологических реагентов и катализаторов. Так, природные белки состоят из левых оптических изомеров аминокислот и поэтому искусственно синтезированные правые аминокислоты организмом не усваиваются; дрожжи сбраживают лишь правые изомеры сахаров, не затрагивая левые и т.д. Общее правило состоит в том, что энантиомеры проявляют идентичные свойства в симметричном (ахиральном) окружении, а в несимметричном (хиральном) окружении их свойства могут изменяться, Это свойство используется в асимметрическом синтезе и катализе. Смесь равных количеств энантиомеров, хотя и состоит из хиральных молекул, оптически неактивна, т.к. одинаковое по величине и противоположное по знаку вращение взаимно компенсируется. Такие смеси называют рацемическими смесями или рацематами. В газообразном состоянии, жидкой фазе и в растворах свойства рацематов обычно совпадают со свойствами чистых энантиомеров, однако в твердом состоянии такие свойства, как температура плавления, теплота плавления, растворимость, обычно отличаются. 1.2 Физические причины оптической активностиВ ахиральной среде два энантиомера имеют одинаковые химические и физические свойства, но их легко отличить друг от друга по специфическому взаимодействию со светом. Один из энантиомеров вращает плоскость поляризации линейнополяризованного (плоскополяризованного) света вправо, а другой энантиомер - на точно такой же угол влево. 1.2 а. Феноменологическая модельФеноменологическую модель оптической активности предложил Френель еще в 1823 г. Она основана на волновой теории света и с позиций современной науки не является достаточно строгой. Тем не менее, эта модель дает очень наглядное представление о причинах оптической активности и других явлениях, связанных с поглощением света хиральным веществом, в рамках классической электродинамики, поэтому ее часто используют и в настоящее время. Согласно классическим представлениям, линейнополяризованный (плоскополяризованный) свет характеризуется тем, что векторы составляющих его зависимых от времени электрического (Е) и магнитного (Н) полей осциллируют во взаимно перпендикулярных плоскостях и их изменения носят синусоидальный характер во времени и в пространстве. Плоскополяризованный свет можно рассматривать как комбинацию левого и правого циркулярнополяризованных лучей, движущихся в фазе по отношению друг к другу. Если в начальной точке времени 1 электрические векторы левого и правого циркулярнополяризованных лучей ориентированы вверх, то в точке 2 вектор правого луча ориентирован вправо, а вектор левого луча влево (если смотреть в направлении движения света по оси z). В точке 3 векторы обоих лучей ориентированы вниз, в точке 4 вектор правого луча ориентирован влево, а вектор левого луча вправо, и т.д. Таким образом, правый и левый циркулярнополяризованные лучи обладают соответственно правой и левой спиральностью вращения вектора электрического поля. Сумма этих лучей дает плоскополяризованный луч, в пространственно-временных точках 1,3 и 5 векторы суммируются, а в точках 2 и 4 взаимно уничтожаются. Расстояние между точками 1 и 5 соответствует одному витку правой или левой спирали или длине плоской волны. При попадании света на любую молекулу в прозрачной среде, скорость его замедляется (уменьшение скорости пропорционально показателю преломления среды), так как свет взаимодействует с электронными оболочками молекул. Степень такого взаимодействия зависит от поляризуемости молекулы.
Плоско (линейно) поляризованный световой луч (а), правый (б) и левый (в) циркулярно-поляризованные лучи, (г) - результат взаимодействия электрических векторов лучей (б) и (в), находящихся в фазе. Если среда ахиральна, две циркулярнополяризованные составляющие проходят с одинаковой скоростью (т.е. с одинаковыми показателями преломления для правого и левого лучей). Однако хиральные молекулы проявляют анизотропию поляризуемости, которая зависит от того, левую или правую спиральность имеет циркулярнополяризованный луч. При прохождении через хиральную среду в общем случае неодинаковы не только скорости, но и коэффициенты поглощения левого и правого циркулярнополяризованных компонент плоскополяризованного света. В результате векторы для правого и левого прошедшего через образец лучей будут иметь разную амплитуду, а результирующий вектор будет описывать эллиптическую траекторию. В общем, при прохождении плоскополяризованного света через хиральную среду вектор электрического поля начинает описывать эллипс (эллиптическая поляризация) с повернутой главной осью. Угол вращения уменьшается с увеличением длины волны падающего света. Однако это справедливо лишь для света, длина волны которого больше длины волны максимума поглощения в электронном спектре данного вещества. Изменение оптического вращения при изменении длины волны называется дисперсией оптического вращения (ДОВ). Разность поглощения правой и левой компонент называется круговым дихроизмом (КД). Количественной характеристикой КД служит угол эллиптичности y, величина которого обратно пропорциональна длине волны КД открыт Э. Коттоном в 1911 г. и его часто называют эффектом Коттона. ДОВ и КД вместе называются хирооптическими явлениями; в своей основе они связаны с электронными переходами в хиральном окружении. Эффект Коттона, т.е. превращение плоскополяризованного света в эллиптически поляризованный заметно проявляется главным образом вблизи полос собственного (резонансного) поглощения вещества.
(а) - Взаимодействие сдвинутых по фазе компонентов равной амплитуды, (б) - взаимодействие находящихся в фазе компонентов разной амплитуды, (в) - суммарный результат сдвига по фазе. 1.2 б. Квантовая теорияКвантовую теорию оптической активности построил в 1928 г. бельгийский физик Л. Розенфельд. С позиций современной науки эта теория рассматривается как более строгая. Для объяснения оптической активности оказалось необходимым учитывать взаимодействие электрических и магнитных дипольных моментов, наведенных в молекуле полем проходящей световой волны. 1.2 в. Корпускулярная теорияВ настоящее время возрождается интерес к корпускулярной теории света, которой придерживался еще Ньютон. Частицей света является фотон - реальная элементарная частица. В фотонной теории поляризацию света связывают с поляризацией фотонов, которая обусловлена наличием у этих частиц спина и его определенной направленностью в пространстве. Спиновые квантовые числа - это как бы дополнительные внутренние степени свободы частицы. В отличие от электронов, имеющих спин J = 1/2, спин фотона J = 1. (Это означает, что электроны принадлежат к классу фермионов, для которых справедлив запрет Паули, а фотоны - к классу бозонов, для которых не действует принцип запрета). Согласно квантовой механике, частица со спином J и ненулевой массой покоя имеет (2J + 1) внутренних квантовых состояний, определяющих ее поляризацию, т.е. степень асимметрии частицы в пространстве. Но масса покоя фотона равна нулю, и поэтому число спиновых состояний на единицу меньше, т.е. равно двум (+1 и - 1). Это означает, что возможны лишь две ориентации проекции спина фотона на направление его движения: параллельная и антипараллельная. В таком случае возникает понятие "спиральность частицы". Если проекция спина на направление движения положительна, то говорят, что частица имеет правовинтовую (правую) спиральность, а если отрицательна - левовинтовую (левую) спиральность. Спиральные объекты хиральны, поэтому фотоны являются как бы хиральными частицами. Поскольку фотоны обладают целочисленным спином, в одном и том же состоянии может находится любое число фотонов. Это обусловливает возможность описания электромагнитных взаимодействий с участием большого числа фотонов в рамках классической (а не только квантовой) механики. Циркулярно-поляризованный свет можно рассматривать как поток фотонов, имеющих только правую или только левую спиральность. Плоскополяризованный свет состоит из одинакового количества "левых" и "правых" фотонов. Взаимодействие по-разному поляризованных фотонов с хиральной анизотропной средой происходит неодинаково, что приводит к хироптическим эффектам. Ахиральная молекула не вращает плоскость поляризации света только при определенной ее ориентации по отношению к падающему лучу. Например, ахиральная молекула, имеющая плоскость симметрии, не вращает плоскость поляризации лишь в том случае, если плоскость поляризации совпадает с плоскостью симметрии. Все же остальные молекулы, не ориентированные таким образом, вращают плоскость поляризации, даже не будучи хиральными. Однако в целом образец не вращает, так как в массе молекулы ориентированы беспорядочно, и одни молекулы вращают плоскость поляризации в одном направлении, а другие молекулы, встречающиеся на пути светового луча, вращают ее в противоположную сторону. Таким образом коллектив ахиральных молекул имеет суммарное вращение, равное нулю, хотя каждая молекула может вращать плоскость поляризации. В случае хиральных соединений молекул противоположной ориентации (если это не рацемическая смесь) просто не может существовать, и вращение наблюдается. 2. Хиральные молекулыВ случае простых молекул легко проводится зрительное распознавание несовместимости с зеркальным отображением. Однако многие органические молекулы настолько сложны, что такой способ требует очень развитого пространственного воображения, которым обладают далеко не все. 2.1 Точечные группы симметрииШар самый симметричный объект, его не возможно отразить в зеркале. Он всегда выглядит одинаково. Тетраэдр "менее симметричен", чем шар, поскольку вокруг высоты его нужно повернуть лишь на определенный угол (1200), чтобы он выглядел так же, как до поворота. Вращение вокруг оси является одной из операций симметрии. Операцией симметрии называется действие над объектом, которое приводит к его новой ориентации, неотличимой от исходной и совмещаемой с нею. Каждой операции симметрии соответствует определенный элемент симметрии. Элементом симметрии называется геометрическое место точек, остающихся неподвижными при данной операции симметрии. Основными элементами симметрии являются собственные оси вращения, которые в системе обозначений Шенфлиса имеют символ Cn, где n - порядок оси, означающий, что поворот молекулы на угол 2p /n радиан приводит к структуре, неотличимой от первоначальной, несобственные оси вращения или зеркально-поворотные оси (s n), зеркальные плоскости симметрии (s), делящие молекулу пополам, так, что одна половина является зеркально-симметричной другой половине, центр инверсии (i) и тождественное преобразование (Е). В соответствии с этим операции симметрии делят на поворот оси вокруг оси симметрии Сn, поворот вокруг оси с последующим отражением в плоскости, перпендикулярной этой оси (Sn), отражение в плоскости симметрии s, инверсию в центре симметрии i и операцию идентичности Е. При операции идентичности с молекулой ничего не делают, но эта операция не бессмысленна, т.к. она позволяет включить в единую классификацию как симметричные, так и асимметричные объекты. 2.1 а. Собственная ось симметрииВсе молекулы имеют тривиальную ось С1, поскольку в любом случае вращение на 3600 возвращает молекулу в исходное состояние. Следовательно, операция С1 эквивалентна операции идентичности (С1 є Е). Дихлорметан имеет ось С2, аммиак - ось С3, метан - четыре оси С3, тетрахлорплатинат - ось С4.
2.1 б. Несобственная ось симметрииПростейшая зеркально поворотная ось S1 эквивалентна перпендикулярной ей плоскости симметрии (S1 є s). Примером является молекула хлорфторметана. Зеркально-поворотные оси более высокого порядка (Sn) можно рассматривать как комбинацию вращения на угол 2p /n с последующим отражением в плоскости, перпендикулярной оси вращения. Так, аллен и изображенный ниже изомер 1,2,3,4-тетраметилциклобутана имеет зеркально-поворотную ось S4:
1,2-Дихлор-1,2-дифторэтан обладает осью S2, которая совпадает со связью С-С. Операция S2 эквивалентна инверсии в центр симметрии, который находится посредине связи С-С (S2 є i)
Поскольку у молекул может быть не один, а несколько элементов симметрии, их удобнее классифицировать по точечной группе симметрии. Набор все операций симметрии объекта образует его группу симметрии. Если при всех этих преобразованиях остается неподвижным центр тяжести фигуры, то группа симметрии называется точечной. Известны четыре типа точечных групп симметрии. 2.1 в. Типы точечных групп симметрииК типу 1 относятся точечные группы С1, Сs, Ci, которые не имеют нетривиальных поворотных осей, поэтому их называют неаксиальными. К типу 2 относятся группы с единственной поворотной осью. В группе Cn других элементов симметрии нет, в группе Cnv имеется n вертикальных плоскостей s n, проходящих через ось Cn, а в группе Сnh одна горизонтальная плоскость s h, перпендикулярная оси Сn. Сюда же входит группа Sn, поскольку при наличии зеркально-поворотной оси порядка n обязательно имеется и собственная ось порядка n/2 (C2 у S4, C3 у S6 и т.д.). При нечетном n оси Sn могут быть представлены как комбинации других операций. Для низших порядков S1 є s и S2 є i. Точечные группы типа 3 имеют одну ось Сn и n осей второго порядка, перпендикулярных оси Сn. Такие группы называются диэдральными. Если нет плоскостей симметрии, группа обозначается как Dn, если имеется несколько плоскостей s v (вертикальных) - Dnd, а если еще и горизонтальная плоскость s h, то группа обозначается Dnh. К типу 4 относятся точечные группы, имеющие более чем одну ось порядка выше двух. Такие группы называются кубическими. К ним относятся точечные группы правильных тетраэдра (Td), октаэдра и куба (Oh), икосаэдра и додекаэдра (Ih). Максимальную симметрию имеет шар, который принадлежит предельной группе Kh, включающей все возможные операции симметрии. 2.2 Симметричное определение хиральностиХиральна любая истинно асимметрическая молекула, относящаяся к группе С1, не имеющая никаких элементов симметрии, кроме идентичности (и оси С1, т.к. С1 Таким образом, можно сформулировать симметрийный критерий хиральности: любая молекула, которая не имеет несобственной оси вращения Sn хиральна. Впервые доказательство справедливости данного определения хиральных молекул получено при исследовании изомерных четвертичных аммонийных солей со спирановым атомом азота IV, V, VII и IX. Изомеры IV и V асимметричны (группа C1), изомер VII диссимметричен (группа D2). Поэтому эти три изомера должны быть хиральными. И действительно, они были получены в оптически активной форме. Однако изомер VIII относится к группе S4, т.е. ахирален, и получить его в оптически активной форме нельзя. 2.3 Типы хиральностиМолекулы, содержащие тетраэдрический атом, например, углерода с четырьмя разными заместителями принадлежат к точечной группе С1. Они асимметричны и центральный атом называется асимметрическим атомом.
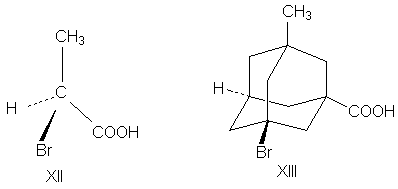
Адамантаны, у третичных атомов углерода которых имеется четыре разных заместителя, хиральны и оптически активны. При сравнении формул симметрия обоих соединений очень похожа. Остов адамантана можно представить как тетраэдр с "изломанными ребрами", он имеет симметрию Td которая переходит в C1, когда все четыре заместителя у третичных атомов углерода разные. У производного адамантана нет асимметрического атома углерода, как в a-бромпропионовой кислоте, но есть центр, находящийся внутри молекулы (центр тяжести незамещенного адамантана). Асимметрический центр - это частный случай более общего понятия хиральный центр. Хиральный центр может иметь не только асимметрические молекулы, но и молекулы симметрии Cn или Dn. Хиральный центр является лишь одним из возможных элементов хиральности. Однако кроме центрального существуют еще и аксиальный, планарный и спиральный типы хиральности. Аксиальной хиральностью обладают молекулы, имеющие хиральную ось. Хиральную ось легко получить, мысленно "растягивая" центр хиральности:
Хиральную ось имеют такие классы молекул, как аллены и дифенилы. В алленах центральный атом углерода sp-типа имеет две взаимно-перпендикулярные p-орбитали, каждая из которых перекрывается с p-орбиталью соседнего атома углерода, в результате чего остающиеся связи концевых атомов углерода располагаются в перпендикулярных плоскостях. Сам аллен хирален, так как имеет зеркально-поворотную ось S4, но несимметрично замещенные аллены типа abС=С=Сab хиральны.
Аллены хиральны только в том случае, если оба концевых атома углерода замещены несимметрично:
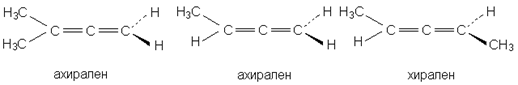
При любом нечетном числе кумулированных двойных связей четыре концевые группы располагаются уже не в разных, а в одной плоскости, например, для 1,2,3-бутатриена:
Такие молекулы ахиральны, но для них наблюдается цис-транс-изомерия. Так, соединение было разделено на оптические изомеры.
Если одну или обе двойные связи симметрично замещенного аллена заменить на циклическую систему, то полученные молекулы будут тоже обладать аксиальной хиральностью, например:
В бифенилах, содержащих четыре объемистые группы в орто-положениях, свободное вращение вокруг центральной связи затруднено из-за стерических препятствий, и поэтому два бензольных кольца не лежат в одной плоскости. По аналогии с алленами, если одно или оба бензольных кольца замещены симметрично, молекула ахиральна; хиральны же молекулы только с двумя несимметрично замещенными кольцами, например:
Изомеры, которые можно разделить только благодаря тому, что вращение вокруг простой связи затруднено, называются атропоизомерами. Иногда для предотвращения свободного вращения в бифенилах достаточно трех и даже двух объемистых заместителей в орто-положениях. Так, удалось разделить на энантиомеры бифенил-2,2-дисульфокислоту (XV). В соединении XVI свободное вращение полностью не заторможено, и, хотя его можно получить в оптически активной форме, при растворении в этаноле оно быстро рацемизуется (наполовину за 9 мин. при 250).
Для некоторых хиральных молекул определяющим структурным элементом является не центр, не ось, а плоскость. Простейшую модель планарной хиральности легко сконструировать из любой плоской фигуры, не имеющей осей симметрии, лежащих в этой плоскости, и отдельной точки вне плоскости. Наиболее изучены планарно-хиральные производные ферроцена (XVII). Другими примерами являются ареновые комплексы хромтрикарбонила (XVIII), а также соединения XIX и XX.
Спиральная хиральность обусловлена спиральной формой молекулы. Спираль может быть закручена влево или вправо, давая энантиомерные спирали. Например, в гексагелицене одна часть молекулы из-за пространственных препятствий вынуждена располагаться над другой.
3. Номенклатура энантиомеровОпределение конфигурации-это экспериментальная работа, выполняемая химическими и физическими методами с целью установить, какая из двух зеркальных пространственных моделей отвечает правовращающему энантиомеру, а какая - левовращающему. При полной определенности самой конфигурации (пространственной модели) вопрос об ее обозначении может решаться по-разному. 3.1 По конфигурации: R - и SСистема R/S - наиболее важная номенклатурная система для характеристики энантиомеров. По этой системе, центр хиральности называется R или S в соответствии с системой, по которой каждое замещающее звено наделяется приоритетом в соответствии с правилами Кана-Инголда-Прелога, основываясь на атомном номере. Если центр ориентирован так, что низший из возможных четырёх направлен от наблюдателя, наблюдатель увидит два возможных варианта: если приоритет оставшихся трёх замещающих групп уменьшается по часовой стрелке, название даётся R (Rectus), если уменьшается против часовой стрелки, то S (Sinister). Эта система маркирует каждый хиральный центр молекулы (и также имеет распространение на хиральные молекулы, не затрагивая хиральных центров). Несмотря на это, она более обобщённа, чем система D/L, и может, например, наименовать изомер, в котором (R,R) - группа расположена напротив (R,S) - диастереомер. У системы R/S нет отношения к (+/-) - системе. R-изомер может быть правоповоротным, так и левоповоротным, в зависимости от фактических замещающих групп. У системы R/S нет и отношения к D/L системе. По этой причине система D/L остаётся в повседневном использовании
3.2 По оптической активности: +/-Энантиомер именуется по направлению света, в котором вращает плоскость поляризованного света. Если вращение происходит по часовой стрелке (по отношению к наблюдателю, к которому направляется свет), то в названии энантиомера отмечается (+). Его зеркальный образ именуется (-). (+) - и (-) - изомеры также определяются как в - и L соответственно (от англ. Dextrorotatory - правоповоротный и Levorotatory - левоповоротный). 3.3 По конфигурации: в - и L-Оптический изомер может быть назван по пространственной конфигурации его атомов. Система D/L делает это, опираясь на молекулу глицераля. Сам по себе глицераль хирален, и два его изомера именуются в и L. С глицералем можно провести определённые химические манипуляции без изменения конфигурации, и его историческое использование с этой целью (в совокупности с удобством его использования как одной из наименьших широко используемых хиральных молекул) вылилось в его использование в номенклатуре. В этой системе составные части называются по аналогии с глицералем, который, в общем, производит недвусмысленные обозначения, да к тому же и легче всего увидеть в маленьких биомолекулах, похожих на глицераль. Маркировка D/L не относится к (+) / (-) никаким образом; она не указывает, какой энантиомер правоповоротный, какой - левоповоротный. Однако она сообщает, что стереохимия соединений имеет отношение к тому, что из право - или левоповоротного энантиомеров глицераля правоповоротный будет D-изомером. Общая закономерность для определения D/L изомерии аминокислот называется правилом “CORN”. Группы COOH, R, NH2 и H (где R - отличная от других углеродная цепь) выстраиваются вокруг атома углерода хирального центра. Когда смотреть так, чтобы атом водорода был направлен вдаль от наблюдателя, если эти группы расположены по часовой стрелке вокруг атома углерода, то это D-форма. Если против часовой, то L-форма.
4. Методы определения конфигурации4.1 Определение абсолютной конфигурацииДля определения абсолютной конфигурации применяются два метода: экспериментальное исследование аномальной дифракции рентгеновских лучей на ядрах тяжелых атомов и теоретический расчет величины оптического вращения. 4.1 а. Дифракция рентгеновских лучейБлагодаря тому, что рентгеновские лучи при прохождении через кристаллы дают дифракционную картину, метод рентгено-структурного анализа (РСА) широко используется для установления строения химических соединений. Когда дифракция происходит на электронных оболочках легких атомов (C,H,N,O,F,Cl), характер наблюдаемой интерференциальной картины определяется только наличием самих ядер, но не их природой. Это объясняется тем, что легкие атомы лишь рассеивают рентгеновские лучи, но не поглощают их, и поэтому в ходе эксперимента не происходит изменения фазы рассеянного излучения. Тяжелые атомы не только рассеивают, но и поглощают рентгеновские лучи в определенных областях кривой поглощения. Если длина волны падающего излучения совпадает с начальным слабо поглощающим участком этой кривой, то наблюдается не только обычная дифракция, но также и некоторый сдвиг по фазе рассеянного излучения, обусловленный тем, что часть его поглощается. Это явление называется аномальным рассеянием рентгеновских лучей. При наличии лишь легких атомов РСА позволяет определить межъядерные расстояния между связанными и несвязанными атомами и на их основе сделать выводы о строении данной молекулы и о наличии в ней хиральных элементов. В этом случае различить энантиомеры нельзя. Однако при наличии тяжелых атомов характер аномального рассеяния зависит не только от расстояния между атомами, но и от относительного расположения в пространстве. Явление аномальной дифракции рентгеновских лучей позволяет непосредственно определить абсолютные конфигурации молекул, содержащих тяжелые атомы, а также молекул, в которые тяжелые атомы могут быть введены в качестве специальных меток. Впервые такой анализ был проведен Бейфутом в 1951 г. В настоящее время с помощью РСА определена абсолютная конфигурация нескольких сотен соединений. 4.1 б. Теоретический расчет оптического вращенияВ 1952 г был опубликован квантово-химический расчет оптического вращения знантиомеров на примере транс-2,3-эпоксибутана (XXX). Конфигурация этого эпоксида может быть скоррелирована с конфигурацией винной кислоты и далее с глицериновым альдегидом. При этом снова обнаружилось, что ранее произвольно выбранная стереоформула D-глицеринового альдегида совершенно правильна и нет необходимости изменять принятое в литературе в течение многих лет изображение этой конфигурации.
4.2 Определение относительной конфигурацииПри определении относительной конфигурации соединение с неизвестной конфигурацией соотносят с другим соединением, конфигурация которого уже известна. 4.2 а. Химическая корреляцияПервая группа методов связана с превращением соединения с неизвестной конфигурацией в соединение с известной конфигурацией или образованием неизвестной конфигурации из известной без нарушения хирального элемента, например, хирального центра. Поскольку в ходе превращения хиральный центр не затрагивается, очевидно, что продукт должен иметь ту же конфигурацию, что и исходное соединение. При этом вовсе не обязательно, что если неизвестное соединение относится к (R) - ряду, то и известное будет иметь (R) - конфигурацию. Например, при восстановлении (R) - 1-бром-2-бутанола в 2-бутанол, не затрагивающем хиральный центр, продуктом будет (S) - изомер несмотря на то, что его конфигурация не изменилась. Это связано с тем, что группа СH3CH2 определению младше группы BrCH2, но старше группы СН3.
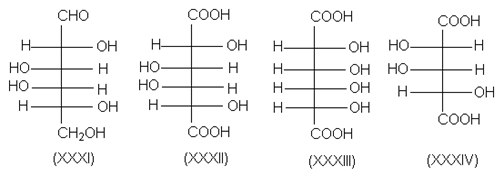
Одним из многих примеров химической корреляции является установление относительной конфигурации D-галактозы (XXXI) путем ее окисления. Поскольку этот процесс приводит к образованию оптически неактивной дикарбоновой кислоты, относительная конфигурация ее четырех хиральных центров может соответствовать или структуре XXXII, или структуре XXXIII. Но дикарбоновая кислота (XXXIV), полученная из галактозы путем окислительного отщепления альдегидного атома углерода, оптически активна. Следовательно, D-галактоза имеет относительную конфигурациию, показанную формулой XXXI.
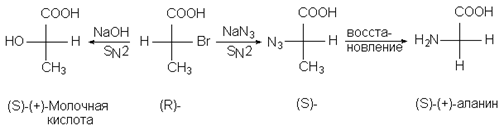
Подобным путем можно выяснить лишь относительную конфигурацию исследуемых молекул, но не их абсолютные конфигурации. Вторая группа методов химической корреляции основана на превращении при хиральном центре, механизм которого точно известен. Так, реакция SN2 происходит с обращением (инверсией) конфигурации реакционного центра. С помощью последовательности таких реакций конфигурация (+) - молочной кислоты была скоррелирована с конфигурацией (S) - (+) - аланина.
К третьей группе относятся биохимические методы. В ряду одного класса соединений, например, аминокислот, определенный фермент атакует молекулы только одной конфигурации. Если какой-то фермент, скажем, атакует только (S) - аминокислоты, не трогая (R) - форму, и это экспериментально установлено на ряде примеров, то еще одна аминокислота, подвергающаяся действию того же фермента, должна принадлежать к (S) - ряду. 4.2 б. Установление относительной конфигурации с помощью физических методовНаиболее широко используют хироптические методы (ДОВ и КД) и спектроскопию ЯМР. Использования хироптических методов для установления конфигурации заключается в сравнении параметров ДОВ и КД в сериях похожих соединений. Эксперимент показал, что знаки эффекта Коттона для этих двух соединений противоположны, но форма и интенсивность спектральных кривых одинакова. Другими словами, кривые ДОВ и КД зеркально-симметричны, и следовательно соединения XXXV и XXXVI можно рассматривать как квазиэнантиомеры в хироптическом (но не в истинно структурном) смысле термина. В приведенном примере Уф - поглощение обусловлено карбонильным хромофором, который ахирален. Тем не менее, наличие хирального окружения оказывает хиральное возмущающее действие на электронный переход группы С=О, позволяя установить относительные конфигурации. При определении относительных конфигураций методом ЯМР обычно используют химические сдвиги и константы спин-спинового взаимодействия. Так, например, в 1,3-дитиане (XVII) экваториальные атомы водорода в положении 2 имеют значительно более высокий химический сдвиг, чем в аксиальном положении, на основании чего легко определить конфигурацию 2-замещенных дитианов.
Константы спин-спинового взаимодействия (J) у вицинальных протонов в этановом фрагменте коррелируют с величинами соответствующих двугранных углов j:
На этом основании можно определить конфигурацию, но только в рядах структурно-родственных соединений, так как величина J зависит также и от природы заместителей. Еще один способ основан на явлении изменения химических сдвигов под влиянием лантанидных комплексов, которые называются сдвигающими реагентами. Известно, что шестикоординационные хелатные комплексы некоторых парамагнитных лантанидов (например, b - дикетонат европия XXXVIII) могут увеличивать координационное число до 8 путем образования неустойчивых ассоциатов с полярными электронно-донорными группыми типа C=O, OH, NH2 и др. Это приводит к сильному изменению величины химсдвигов ядер близко расположенных к координирующему атому. Таким путем можно, например, отличить экзо - и эндо-изомеры борнеола (XXXIX).
Конфигурацию гомологов можно определить просто по знаку оптического вращения. В гомологических рядах вращение обычно меняется постепенно и в одном направлении, поэтому, если известна конфигурация достаточного числа членов данного ряда, конфигурацию остальных можно установить экстраполяцией. 5. Методы разделения энантиомеровОперации разделения рацемических смесей на составляющие их оптически активные компоненты называются расщеплением. Отношение экспериментально наблюдаемого удельного вращения вещества, полученного путем расщепления, к удельному (абсолютному вращению чистого энантиомера называется оптической чистотой (Р). Тождественными оптической чистоте являются понятия энантиомерной чистоты или энантиомерного избытка (э. и.).
где Е - мольная доля энантиомера, находящегося в избытке, Е* - мольная доля другого энантиомера. Любой процесс получения оптически активного вещества из оптически неактивного предшественника, в том числе и расщепление рацемических смесей, называется оптической активацией. Общим принципом всех процессов оптической активации является создание в той или иной форме диастереомерных взаимодействий. 5.1 Расщепление через диастереомерыЭтот метод до настоящего времени использовался наиболее часто. Если рацемическое соединение содержит карбоксильную группу, то можно получить соль с оптически активным основанием. Если же рацемат содержит аминогруппу, то можно получить соль с оптически активной кислотой. Допустим, что оптически активный реагент (в данном случае основание или кислота) имеет (S) - конфигурацию. Тогда образующиеся соли будут смесью (R) - и (S) - диастереомеров, и в отличие от энантиомеров их свойства будут уже различаться.
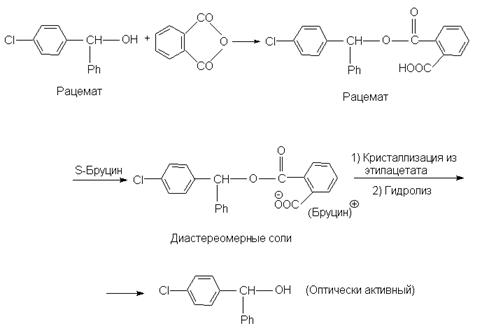
На практике чаще всего применяют кристаллизацию, используют различие в растворимости двух диастереомеров. В настоящее время все чаще применяют хроматографические методы. На последней стадии из соли выделяют знантиомер. Для разделения рацемических кислотных соединений применяют природные оптически активные основания, которые называются алкалоидами, например, бруцин, эфедрин, стрихнин, хинин, цинхонин, морфин и др. После проведения разделения их регенерируют и используют снова. Однако эти вещества сильно токсичны и поэтому их стремятся заменить синтетическими оптически активными аминами, например, a - фенилэтиламином. Например, таким путем расщепляется рацемическая 3-метил-2-фенилбутановая кислота.
Для разделения рацемических основных соединений применяют оптически активные кислоты: винную, миндальную, аспарагиновую (аминоянтарную), глутаминовую (a - аминоглутаровую), камфорсульфоновую и др. Если молекула не содержит кислотной или основной группировки, то ее можно сначала ввести, а затем после разделения на энантиомеры снять, например,
Диастереомеры могут образовываться не только в результате взаимодействий кислот и оснований Бренстеда, но также и в реакциях, в которых взаимодействуют кислоты и основания Льюиса. Так, при расщеплении ароматических соединений, в состав которых не входит ни кислотные, ни основные группировки (например, хиральных нафтиловых эфиров), может быть использована их способность образовывать p - комплексы с нитрофлуореном. Для этой цели используют реагент (XLI), в котором элекктроноакцепторные тетранитрофлуореноноксимная группа придает ей способность к комплексообразованию с электронодонорными ароматическими кольцами, а фрагмент энантиомерной молочной кислоты обеспечивает реагенту в целом оптическую активность. Другим примером является расщепление транс-циклооктена путем образования комплекса с солью двухвалентной платины (кислота Льюиса), вторым лигандом у которой является молекула (R) - a - фенилэтиламина (XLII).
5.2 Хроматографическое расщеплениеЕсли рацемичеcкую смесь хроматографировать на колонке, заполненной хиральными веществами, энантиомеры должны проходить с разными скоростями и, следовательно, их можно разделить. Таким путем, например, миндальную кислоту разделяют на колонке, заполненной крахмалом. Можно использовать бумажную, колоночную, газовую и жидкостную хроматографию. 5.3 Механическое расщеплениеВ случае рацемической натрийаммониевой соли винной кислоты энантиомеры при температуре ниже 270 (температура очень важна) кристаллизуются раздельно: в одном кристалле собираются (+) - изомеры, а в другом (-) - изомеры. Такие кристаллы отличаются друг от друга зеркальностью формы, и их можно разделить с помощью пинцета и микроскопа. Именно таким путем Л. Пастер в 1848 г. впервые доказал, что рацемическая винная кислота в действительности представляет собой смесь (+) - и (-) - изомеров. Однако такого рода кристаллизация свойственна лишь немногим веществам. Описано, например, расщепление гептагелицена (смесь спирально сочлененных бензольных колец; аналог гексагелицена - ). Один из энантиомеров этого соединения, имеющий необычно высокое оптическое вращение ([a] D20= +62000) спонтанно выкристаллизовывается из бензола. При аналогичном расщеплении 5-метил-3,3-диэтил-2,4-пиперидиндиона (XLIII) было взято 20 кг рацемата и после 400 перекристаллизаций получено всего 3 г оптически чистого правовращающего изомера. Одним из немногих соединений, которые можно разделить пинцетом по методу Пастера является 1,1,-динафтил (XLIV). При нагревании рацемата при 76-1500 происходит фазовое изменение с образованием лево - и правовращающих кристаллов.
5.4 Ферментативное расщеплениеДовольно часто для получения оптически активных веществ из рацематов используют ферменты, которые обладают высокой стереоспецифичностью действия. Наибольшее значение метод приобрел для стереоспецифического гидролиза N-ациламинокислот. Под действием фермента ацилазы на рацемическую N - ацетиламинокислоту L-изомер гидролизуется в 1000 раз быстрее D-изомера, и после окончания ферментативной реакции легко можно разделить L-аминокислоту и D-ацетиламинокислоту. 5.5 Установление оптической чистотыВ большинстве случаев при расщеплении рацематов получаются энантиомеры, не имеющие 100% -ной оптической чистоты. Для установления содержания в них второго энантиомера применяют по сути дела те же методы, что и для расщепления, с той лишь разницей, что в данном случае образующиеся диастереомерные комплексы не разделяют, а тем или иным способом определяют их концентрацию. Относительные концентрации диастереомеров можно определить любым способом, например, с помощью ГЖХ или ЯМР-спектроскопии. ЗаключениеОпти́ческая изомери́я - разновидность пространственной изомерии, являющаяся прямым следствием хиральности молекул, проявляется способностью некоторых веществ поворачивать плоскость поляризованного луча в противоположные стороны. Оптическая изомерия свойственна молекулам органических веществ, не имеющим плоскости симметрии, которые относятся друг к другу как предмет к своему зеркальному отражению. Два стереоизомера, относящиеся друг к другу как предмет к своему зеркальному отражению, не совместимому с оригиналом, называются энантиомерами, и каждая из этих структур является хиральной. Существование двух энантиомеров (хиральность) обусловлено атомом, имеющим различные заместители. Такой асимметрический атом называют стереоцентром или стереогенным центром. Применяются также названия хиральный или асимметрический центр. Смесь равных количеств обоих энантиомеров называется рацемической формой. Некоторые характеристики энантиомеров, например растворимость и реакционная способность, одинаковы только при ахиральном окружении, если же энантиомер окружен хиральными молекулами, реакционная способность двух энантиомеров будет различаться. Энантиомеры различаются также при прохождении луча плоско поляризованного света через их растворы. Для каждой пары энантиомеров луч отклоняется на один и тот же угол, но в разные стороны (направо или налево), что обозначается знаками "+" и "-" или в и l. По этой причине стереоизомеры такого типа иногда называют оптическими изомерами. В обычных химических реакциях, приводящих к образованию энантиомеров, получаются их равные количества (рацемическая форма). Рацемическая смесь не обладает оптической активностью. Если же химическая реакция проводится в хиральной среде или в присутствии хирального катализатора, то получают продукты с преобладанием (иногда полностью) одного энантиомера. Наличие оптической изомерии может быть обусловлено также наличием стереогенной оси или плоскости. Если молекула содержит более одного стереогенного центра, то число оптических изомеров определяют по формуле 2n, где n - число стереогенных центров. Стереоизомеры, не являющиеся энантиомерами, называются диастереомерами. Операции разделения рацемических смесей на составляющие их оптически активные компоненты называются расщеплением. Отношение экспериментально наблюдаемого удельного вращения вещества, полученного путем расщепления, к удельному (абсолютному вращению чистого энантиомера называется оптической чистотой (Р). Тождественными оптической чистоте являются понятия энантиомерной чистоты или энантиомерного избытка. Существует несколько способов разделения: расщепление через диастереомеры, хроматографическое расщепление, механическое расщепление, ферментативное расщепление и установление оптической чистоты. Литература1. Аблесимов Н.Е. Синопсис химии: Справочно-учебное пособие по общей химии. - Хабаровск: Изд-во ДВГУПС, 2005. 2. Бакстон Ш., Робертс С. введение в стереохимию органических соединений. - М.: Мир, 2005. 3. Вайлен С. Дойл М. Илиел Э. Бином. Лаборатория знаний - 2007. 4. Ельницкий А.П. Номенклатура органических соединений. Мн.: Сэр-Вит, 2003 5. Илиэл Э. Основы стереохимии. М.: Мир, 1971. 6. Ким А. М Органическая химия: Учеб. пособие. - 3-е изд., испр. и доп. - Новосибирск: Сиб. унив. изд-во, 2002 7. Ногради М. Стереохимия. - М.: Мир, 1984. 8. Папулов Ю.Г. Статистическая стереохимия и конформационный анализ. Калинин: КГУ, 1978. 9. Потапов В.М., Стереохимия, М., 2009. 10. Основы стереохимии (пер. с англ. Демьянович В. М.) Изд.2-е 11. Реутов О.А., А.Л. Курц, К.П. Бутин "Органическая химия. Углубленный курс " 1999. 12. Реутов О.А., А.Л. Курц, К.П. Бутин "Органическая химия" - М., 2007 - Ч.2 13. Соколов В.И. Введение в теоретическую стереохимию. - М.: Наука, 1982. 14. Травень В.Ф., Баберкина Е.П., Сафронова О.Б., Шкилькова В.Н. - Стереохимия. Учебное пособие - Москва: РХТУ, 1999. 15. Черних В. П, та ін. Органічна хімія / В.П. Черних, Б, С. Зименковський, І.С. Гриценко: Підручник для фарм. вузів і факультетів. У 3 кн.: Кн.1. Основи будови органічних сполук. - Вид-во "Основа" при Харк. ун-ті. 2000 р. |