| Учёт неидеальности растворов в кинетических исследованиях
Поскольку константа скорости элементарной стадии включает коэффициенты активности реагентов и переходного состояния (активированного комплекса), а константы равновесия комплексов, входящих в материальный баланс по катализатору, – коэффициенты активности участников соответствующего равновесия, важно сохранить при варьировании состава раствора все γi постоянными или учитывать их изменение. Так, например, в случае механизма (16)
 (16) (16)
с лимитирующей второй стадией
 (17) (17)
где 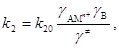  k20 и K10 – константы скорости и равновесия, не зависящие от состава раствора и определяемые природой растворителя. k20 и K10 – константы скорости и равновесия, не зависящие от состава раствора и определяемые природой растворителя.
Естественно, при определении вида уравнения в ходе однофакторного или многофакторного эксперимента необходимо, чтобы значения k2 и K1 не менялись при варьировании [A], [B] и [ ], т.е. надо сохранить постоянными факторы ], т.е. надо сохранить постоянными факторы
 и и 
В водных растворах электролитов используют принцип постоянства ионной силы I или ионной среды (и.с.). Величина I определяется уравнением  где ci и zi – концентрация иона и его заряд. Коэффициенты активности заряженных частиц согласно теории Дебая-Гюккеля зависят от I: где ci и zi – концентрация иона и его заряд. Коэффициенты активности заряженных частиц согласно теории Дебая-Гюккеля зависят от I:
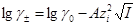 первое приближение (18) первое приближение (18)
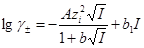 третье приближение (19) третье приближение (19)
Поддерживая в ходе эксперимента высокую и постоянную величину I (или и.с.) с помощью добавок электролитов (и.с. – концентрация инертного сильного электролита), сохраняют постоянство всех γi (F1 и F2).
Например, при исследовании кинетики окисления С2Н4 в растворах комплексов PdCl42– в присутствии ионов Cl– и H3O+ поддерживали постоянной сумму концентраций электролитов при концентрации PdCl42– ≤ 0.2 M.
LiCl – LiClO4 – HCl – HСlO4 = 3M (I = 3, и.с. =3)
Концентрации H3O+ и Cl– варьировали, заменяя LiClO4 на HСlO4 и LiClO4 на LiCl, соответственно.
В ряде случаев удаётся измерять величины, пропорциональные активностям ионов металла и комплексов МLn, что позволяет при поддержании высокого анионного или катионного фона варьировать концентрации MLn в очень широком диапазоне. Так, например, изучалась каталитическая система CuCl – NH4Cl – H2O в разнообразных каталитических процессах. При высокой величине катионного фона ([NH4+]=6 –20 моль/1000г. Н2О) коэффициенты активности Cl–, CumCln(n–m)–, H3O+ и всех интермедиатов оставались постоянными в очень широком интервале концентраций соли меди(I).
Изменение коэффициентов активности сильных кислот в большом интервале концентрации кислот описываются эмпирическим уравнением (20)
 (20) (20)
где mS – моляльная концентрация кислоты или сильного электролита.
Таким же уравнением описывается (в определённом интервале концентраций) и коэффициент активности молекул неэлектролитов. Так, например, известное уравнение Сеченова определяет растворимость органических молекул и различных газов в водных растворах электролитов
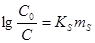 , (21) , (21)
где С0 – растворимость в чистой воде. Это эмпирическое уравнение отражает влияние электролита не только на γ растворённой молекулы, но и на активность воды. Рассмотрим процесс растворения молекулы В как химическую реакцию гидратации В.
 (22) (22)
Тогда для термодинамической константы равновесия в растворе соли получим
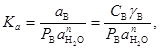 (23) (23)
для чистой воды
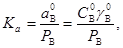 (24) (24)
Из (24) и (23) для РВ = 1 атм. запишем уравнение для растворимости газа В
 (25) (25)
 (26) (26)
Из уравнения (26) (уравнение Флида) видно, член KSmS в уравнении Сеченова (21) отражает влияние соли на γВ и  . Активность всегда меньше . Активность всегда меньше  , и поэтому рост второго члена приводит к падению СВ, а коэффициент γВ может сложным образом меняться от концентрации электролита (и понижаться, и увеличиваться), поэтому и наблюдаются эффекты ”высаливания” и “всаливания” вещества В. , и поэтому рост второго члена приводит к падению СВ, а коэффициент γВ может сложным образом меняться от концентрации электролита (и понижаться, и увеличиваться), поэтому и наблюдаются эффекты ”высаливания” и “всаливания” вещества В.
Влияние электролита на растворимость реагента важно учитывать в кинетических экспериментах в растворах. Так, например, исследование влияния [HCl] на скорость гидрохлорирования ацетилена в системе C2H5OH – HCl – K2PtCl4 показало, что следует учитывать зависимость концентрации ацетилена в растворе от концентрации HCl (эффект ”высаливания”). При этом
 (27) (27)
При варьировании концентрации HCl в большом интервале (1 ÷ 10М) молярная концентрация этанола (растворителя) также заметно меняется.
Исследование каталитических реакций в концентрированных растворах кислот в воде и органических растворителях проводят с использованием различных функций кислотности растворов, например, функции Гаммета Н0:
 (28) (28)
где h0 – кислотность, γВ и γВН+ – коэффициенты активности непротонизованной и протонизованной форм индикатора,  , , 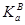 – константы диссоциации протонизованной формы индикаторов. Величина функции кислотности также зависит от концентрации электролита (I). Эта зависимость описывается уравнением Моисеева-Флида – константы диссоциации протонизованной формы индикаторов. Величина функции кислотности также зависит от концентрации электролита (I). Эта зависимость описывается уравнением Моисеева-Флида
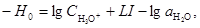 (29) (29)
где L – коэффициент, зависящий от природы кислоты и электролита.
При исследовании кинетики реакций в растворах металлокомплексов следует учитывать влияние состава раствора на величину закомплексованности катализатора. Изменение концентраций катализатора и реагентов не должно влиять на величину  . Это облегчает задачу определения вида кинетического уравнения. В противном случае необходимо знать структуру FM, значения констант равновесия и учитывать изменения величины закомплексованности при варьировании концентраций. Так, например, при изучении зависимости скорости реакции гидрохлорирования ацетилена в системе HgCl2 – HCl – H2O от [HgCl2] было обнаружено, что порядок по [HgCl2] выше единицы . Это облегчает задачу определения вида кинетического уравнения. В противном случае необходимо знать структуру FM, значения констант равновесия и учитывать изменения величины закомплексованности при варьировании концентраций. Так, например, при изучении зависимости скорости реакции гидрохлорирования ацетилена в системе HgCl2 – HCl – H2O от [HgCl2] было обнаружено, что порядок по [HgCl2] выше единицы
R = k[HgCl2]n n > 1.5
хотя никаких димерных комплексов в растворе нет. Высокий порядок объясняется понижением концентрации Cl–, входящего в закомплексованность HgCl2
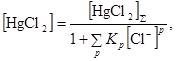 (30) (30)
где р = 1 и 2, а также повышением h0 (уравнение (29)) при появлении в растворе заряженных ионов 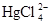 . .
Более сложная задача – изучение кинетики реакций в органических растворителях. В зависимости от полярности растворителя и природы электролита в таких системах образуются различные ассоциаты, ионные пары, ионные тройники и свободные ионы. Поэтому необходимо подходить к каждой системе индивидуально, используя информацию о состоянии солей и комплексов металлов в растворе. Например, Pd(OAc)2 в бензоле и в уксусной кислоте находится в форме тримера, Pd3(OAc)6. Добавление NaOAc приводит к разрушению тримера и к образованию димерного и мономерного комплексов Na2Pd2(OAc)6 и Na2Pd (OAc)4 без образования свободных ионов. Вместе с тем очевидно, что при наличии высоких постоянных концентраций электролита и в органических растворителях (KI – метанол, LiCl – ROH) можно найти условия, при которых изменение концентрации комплекса металла – катализатора не приведёт к изменению функции закомплексованности. Наличие высокой и постоянной концентрации электролита в органических растворителях позволяет также поддерживать постоянными концентрацию ионных пар и внешнесферных комплексов в растворе при варьировании концентраций комплексов металла и реагентов.
“Идеальные” и “неидеальные” поверхности в гетерогенном
катализе
Однородной поверхностью (Лэнгмюр) называют такую поверхность, все центры которой: (1) имеют одинаковую величину константы равновесия адсорбции вещества А ( ), независящую от степени заполнения поверхности Θi
(одинаковые ΔGa
и ΔHa
адсорбции), (2) одинаково способны взаимодействовать со всеми молекулами – участниками реакции (один центр – одна молекула), а адсорбированные частицы не влияют на адсорбцию друг друга. В ходе лэнгмюровской адсорбции возникает идеальный адсорбированный слой. На таких поверхностях адсорбция реагентов (образование поверхностных комплексов) описывается изотермой Лэнгмюра (31) ), независящую от степени заполнения поверхности Θi
(одинаковые ΔGa
и ΔHa
адсорбции), (2) одинаково способны взаимодействовать со всеми молекулами – участниками реакции (один центр – одна молекула), а адсорбированные частицы не влияют на адсорбцию друг друга. В ходе лэнгмюровской адсорбции возникает идеальный адсорбированный слой. На таких поверхностях адсорбция реагентов (образование поверхностных комплексов) описывается изотермой Лэнгмюра (31)
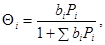 (31) (31)
где bi
– константа равновесия адсорбции для i-того вещества, Pi
– парциальное давление i-того реагента.
В рамках теории Лэнгмюра используют также приближение равномерно- неоднородной поверхности, когда на поверхности есть несколько сортов центров (2, 3), обладающих специфическим сродством к различным молекулам (адсорбатам), но в пределах каждого сорта центров выполняются закономерности лэнгмюровской адсорбции. Тогда при адсорбции, например, Н2
и С6
Н6
получим
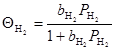 и и 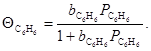
При адсорбции смеси молекул (А и В), когда адсорбция А требует несколько соседних центров, а адсорбция молекулы В протекает на одном центре, простые представления Лэнгмюра уже не работают даже, если поверхность ведёт себя как идеальная адсорбционная поверхность. Требуется более сложный аппарат теории вероятности для описания многоцентровой адсорбции.
Несмотря на то, что наличие идеальной поверхности для большинства твёрдых адсорбентов и катализаторов представляется маловероятным и многочисленные эксперименты подтверждают скорее неоднородность поверхности, представления Лэнгмюра нашли широкое применение для описания кинетики промышленных процессов (кинетика Лэнгмюра-Хиншельвуда). Предполагают, что это связано с тем, что в промышленных условиях большинство процессов протекает в области средних и высоких заполнений поверхности, когда наличие наиболее активных центров (с другими значениями ΔGa
и ΔHa
) уже не чувствуется. Скорость поверхностной реакции, являющейся лимитирующей стадией,

или в виде

запишется по Лэнгмюру-Хиншельвуду уравнением (32)
 (32) (32)
Теория катализа на неоднородных поверхностях развита Тёмкиным, Фрумкиным и Рогинским. Изучение адсорбции различных молекул на металлах и окислах показало, что во многих случаях теплота адсорбции q (q = –ΔHa
) и ΔGa
падает по мере заполнения поверхности. На таких поверхностях изотерма Лэнгмюра не выполняется и появляются логарифмическая изотерма Шлыгина-Фрумкина-Темкина (33) и степенная изотерма Фрейндлиха (34):
 (33) (33)
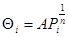 (n > 1) (34) (n > 1) (34)
Рассматривают 2 модели неоднородности – биографическую неоднородность ( на катализаторе до адсорбции есть центры разного сорта) и индуцированную неоднородность, которая возникает в процессе адсорбции вследствие изменений свойств свободных центров и взаимного влияния адсорбированных частиц (латеральное взаимодействие).
Рассмотрим описание кинетики простой двухстадийной реакции на биографически неоднородной поверхности. Если на поверхности имеется несколько сортов центров с долей каждого сорта Wк
, но на каждом сорте центров выполняется уравнение Лэнгмюра, получим:
 и и  , ,
где 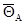 – общая доля поверхности, занятая А. Обозначим величину – общая доля поверхности, занятая А. Обозначим величину  и и 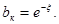 Распределение центров по величинам x описывается производной: Распределение центров по величинам x описывается производной:
 (35) (35)
j(x) – функция распределения центров по величине x. Величину x называют показателем десорбируемости. Установлено, что изотерме Фрейндлиха отвечает функция j(x) (36)
 (36) (36)
а логарифмической изотерме адсорбции – функция j(x) в виде константы
j(x) = const (37)
Количество центров, находящихся в интервале значений от x0
до x, определяют интегрированием выражения (35)
 (38) (38)
 (39) (39)
Если скорость лимитирующей стадии

Равна
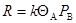
на центрах одного сорта, то на неоднородной поверхности суммарная скорость
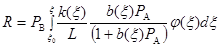 (40) (40)
где величины 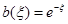 , а для записи , а для записи  используют уравнение Бренстеда ( используют уравнение Бренстеда ( ), связывающее константы скорости стадии с константами её равновесия. ), связывающее константы скорости стадии с константами её равновесия.
Для механизма (41)
  (41) (41)
величина скорости реакции на центрах одного сорта
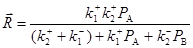 (42) (42)
Запишем для kj
выражения  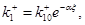  , , 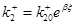 и и  где где 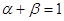 . После интегрирования получим уравнение . После интегрирования получим уравнение
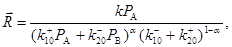 (43) (43)
которое, как легко видеть, сильно отличается от уравнения (42)
В качестве примера кинетики промышленного процесса на неоднородной поверхности приведем знаменитое уравнение синтеза аммиака (М. И. Темкин) на железном катализаторе, которое описывает процесс синтеза NH3
в очень широком интервале давлений (1 – 70 атм.).
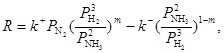 (44) (44)
где m = 0,5 для железного катализатора, k+
/k–
= K – константа равновесия итогового уравнения.
При выводе уравнения принимаем, что поверхность катализатора неоднородна в отношении адсорбции N2
(логарифмическая изотерма), и что диссоциация N2
до атомарного состояния является лимитирующей стадией процесса
Z+N2
 ZN2
ZN2
ZN2
+Z 2ZN 2ZN
2ZN+H2
2ZNH
ZNH+H2
Z+NH3
,
протекающего по итоговому уравнению:
N2
+3H2
2NH3
Статистическое планирование кинетического эксперимента
При планировании кинетического эксперимента важно выбрать статистически обоснованную область изменения параметров , чтобы охватить все возможные сочетания концентраций реагентов и найти такие области концентраций, для которых поведение кинетических уравнений различалось бы наиболее заметно. Например очевидно, что уравнения (45) и (46)
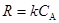 (45) (45)
 (46) (46)
будут различными только при больших концентрациях СА
Параболический характер зависимости R от CA
в уравнение (47), напротив будет заметен в области малых концентраций СА
 (47) (47)
В настоящее время разработан математический аппарат для планирования кинетических экспериментов.
Вопросы для самоконтроля
1) Какие условия должны выполняться при исследовании кинетики гомогенной реакции в системе газ – жидкость в проточном реакторе полного смешения?
2) Перечислить критерии определения внешнекинетического режима проведения гетерогенной каталитической реакции.
3) Перечислить типы идеальных реакторов.
4) Найти уравнение, описывающее зависимость концентрации продукта на выходе из реактора полного смешения от времени контакта для реакции
5) A ® 2B с кинетическим уравнением |RA
| = kCA
при W = W0
.
6) Почему понижение давления переводит процесс в кинетическую область?
7) Что определяет фактор эффективности катализатора?
8) Почему следует поддерживать постоянной ионную силу раствора или ионную среду в кинетических исследованиях?
9) Что произойдет с концентрацией иона H3
O+
в воде, если к раствору уксусной кислоты добавить сильный электролит (например, KCl, LiClO4
)?
10) Какую поверхность твердого вещества можно считать однородной?
11) В каких условиях образуется идеальный адсорбированный слой?
12) Какая функция распределения активных центров по свободным энергиям (или теплотам) адсорбции соответствует логарифмической изотерме адсорбции?
Литература для углубленного изучения
1. Гаммет Л. Основы физической органической химии, М., Мир, 1972, 534 с.
2. Моисеев И.И. p-Комплексы в жидкофазном окислении олефинов, М., Наука, 1970, 240 с.
3. Киперман С.Л. Основы химической кинетики в гетерогенном катализе, М., Химия, 1979.
4. Розовский А.Я. Гетерогенные химические реакции, М., Наука, 1980, 323 с.
5. Одинцов К.Ю., Брук Л.Г., Темкин О.Н. Статистическая обработка результатов кинетических исследований (методическое пособие), М., МИТХТ, 2000, 50 с.
6. Горский В.Г. Планирование кинетических экспериментов, М., Наука, 1984.
|