Элементы статистической термодинамики.
Равновесие закрытой системы в изохорно-изотермических условиях. Макро и микросостояния. Канонический ансамбль. Энтропия и вероятность. Распределение Больцмана. Статистические суммы.
Всякая термодинамическая система состоит из очень большого числа механических объектов. Это коллектив из множества однотипных частиц. Частицы в ней могут быть и микроскопическими – атомно-молекулярного размера, и макроскопическими, подобно частицам взвеси в мутной воде или в пыльном воздухе. Для начала неплохо ещё было бы, чтобы они могли каким-то способом перемешиваться.
Если система пребывает в равновесии, то и перемешивание её механических объектов-подсистем в чём-то похоже на стационарное движение, и систему из равновесия не выводит. Оставаясь в нём, тем не менее, она на уровне микрообъектов всё время пребывает в каких-то изменениях, но все они совместимы с одним этим общим равновесием.
Всё множество способов, которыми реализуется макросостояние, для внешнего лабораторного наблюдателя одинаково. Он не может выделить тех различий, которые неизбежны при перемешивании, и ему незаметен постоянный обмен энергией между частицами.
Все эти изменения, которые не выводят систему из равновесия, состоят в непрерывном перераспределении орбитальных конфигураций в огромном фазовом пространстве, которое представляет из себя не что иное, как множество всех орбиталей, порождаемых всевозможными стационарными движениями всех частиц коллектива, составляющего материальную систему. Конечно же, квантовое фазовое пространство жто дискретное абстрактное математическое множество.
Удивительно, что количество ячеек – орбиталей в нём можно точно вычислить. Но удивительно лишь на первый взгляд...
Для нас важны поступательные (трансляционные), вращательные (ротационные), колебательные (вибрационные), электронные движения и порождённые ими орбитали. Среди них частицы коллектива распределены, они образуют некие мгновенные орбитальные конфигурации, которые постоянно изменяются за счёт всевозможных перескоков частиц между состояниями-орбиталями–уровнями.
Основной механизм, вызывающий эти перераспределения, можно связать с беспорядочными соударениями при броуновском движении.
В свою очередь, каждая конфигурация также порождает множество микросостояний. Коллектив как бы постоянно мигрирует между разными микросостояниями, но в том-то и состоит их отличительная черта, что это никак внешне не сказывается на физически наблюдаемых характеристиках.
С одним-единственным макросостоянием - внешне проявляемым физически наблюдаемым состоянием коллектива - совместимы все эти микросостояния.
Весь набор микросостояний, признаки которых совместимых с макросостоянием, по Гиббсу называется ансамблем, а их число называется термодинамической вероятностью макросостояния.
Наша задача резко облегчена тем, что мы предварительно располагаем такой мощной информацией, какую предоставляет нам формальная химическая термодинамика, и уже можем не моделировать и вычислять термодинамические вероятности. Введённые Гиббсом простые способы и оценки столь универсальны, что остаётся лишь удивляться конкретности и точности прогнозов на их основе.
Нам вполне достаточно даже самых общих представлений о том, что огромное множество дискретных частиц порождают огромное же множество дискретных состояний-уровней, внутри которых все они и пребывают, и распределяются.
Формула Больцмана – вывод (обоснование Планка)
Цель – вывод функции S(W)…
Если система состоит из двух достаточно больших независимых подсистем, то в первом приближении каждую из них и протекающие в них события можно рассматривать как независимые, вводя для них собственные термодинамические вероятности.
Общая термодинамическая вероятность единой системы в таком случае образуется как произведение двух термодинамических вероятностей независимых подсистем.
W=W1W2;
S(W1W2)= S(W1)+ S(W2)
S(W); S(W1); S(W2) - ?
∂W/∂W1=W2; ® ∂W/∂W1=W/W1;
∂W/∂W2=W1; ® ∂W/∂W2=W/W2.
∂S(W)/∂W1= [∂S(W)/∂W][∂W/∂W1]= W2[∂S(W)/∂W] =[W/W 1] [∂S(W)/∂W];
∂S(W)/∂W2= [∂S(W)/∂W][∂W/∂W2]= W1[∂S(W)/∂W] =[W/W2] [∂S(W)/∂W];
W1∂S(W)/∂W1= W1∂S(W1)/∂W1=W[∂S(W)/∂W];
W2∂S(W)/∂W2= W2∂S(W2)/∂W2=W[∂S(W)/∂W];
Результат:
W1∂S(W)/∂W1=W[∂S(W)/∂W]=W2∂S(W)/∂W2 =const;
Для любой подсистемы в общем виде дифференциальное уравнение:
Wi∂S(Wi)/∂Wi=k;
dS(Wi)=kdWi/Wi; ®òdS(Wi)=kòd(lnWi) ; ®S(Wi)=klnWi + lnC; ®
(Wi=1 ® S=0) ® lnC=0; ®S(Wi)=klnWi
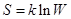
Равновесие в изохорно-изотермической системе.
Каноническое распределение.
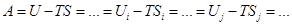


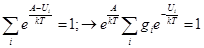

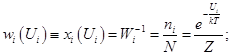
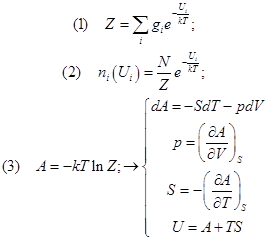
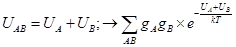

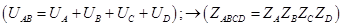



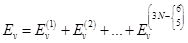

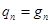

Трансляционная сумма состояний на 1 степень свободы поступательного движения
En= n2 (h2/8mL2)= n2 Bt; "nÎN{1,2,3,…¥} ; gn=1
qt=Sexp[-n2 (Bt/kT)] =
Ротационная сумма состояний на 2 степени свободы вращательного движения (2х ат.мол.)
EL= L(L+1) (ħ2/2mr2)= L(L+1)Br; "LÎZ0{0,1,2,3,…¥}; gL=2L+1
qr2=S(2L+1)exp[-L(L+1) (Br/kT)] =;
Вибрационная сумма состояний на 1 степень свободы колебательного движения
EV= (V+1/2)hn0; "VÎN{1,2,3,…¥}
qV=Sexp[-(V+1/2){hn0/kT}]= ; gV=1
1) 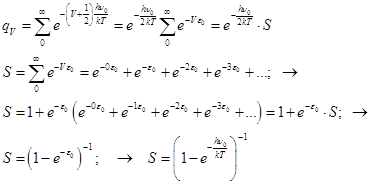
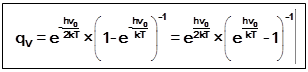
2) 
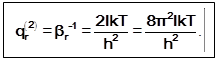
3) 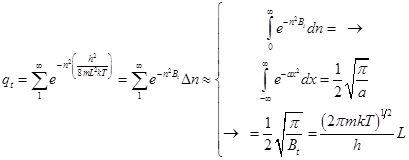
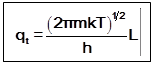
Лекция 17. Статистические суммы молекулярных движений и учёт симметрии. Свободная энергия, энергия Гиббса, уравнение состояния, химический потенциал, стандартизация. Стандартное сродство. Константа равновесия химической газовой реакции.
Началом отсчёта колебательной энергии удобно считать нулевой вибрационный уровень. Это связано с тем, что у простых двухатомных молекул экспериментально именно с основного колебательного уровня определяется энергия диссоциации, именно она является основой всех оценок прочности связи, а нуль вибрационной энергии просто-напросто запрещён принципом Гейзенберга. Он может быть определён только теоретически в качестве гипотетического минимума потенциальной кривой (кривой Морзе - адиабатического потенциала - энергетической кривой основного электронного терма молекулы (одной двухцентровой связи) в приближении Борна-Оппенгеймера).
1) Коррекция колебательной суммы состояний состоит в уточнении способа отсчёта энергии колебаний. Её лучше отсчитывать не от нуля, а от основного уровня (с V=0), которому отвечает остаточная энергия колебаний. Именно с этого уровня должна была бы происходить диссоциация химической связи в гипотетических условиях при абсолютном температурном нуле.
Вибрационные уровни энергии, отсчитываемые от уровня остаточных колебаний и вибрационная статистическая сумма определяются простейшими формулами:
Ev=vh0; → qv(ν0,T)=[1-exp(-hν0/kT)]-1.
Для каждой вибрационной степени свободы записывается своя собственная сумма состояний.
2) Коррекция вращательной суммы состояний состоит в учёте числа симметрии двухатомной гомоядерной молекулы A2:
qr(2)= 8pIkT/h¬s ®qr(2) = 8pIkT/sh
Эта приближённая формула qr(2) пригодна для молекулы линейной формы, у которой имеется две внешние ротационные степени свободы.
Для каждой вращательной степени свободы можно приближённо записать свою собственную сумму состояний qr(1), представляя её в виде квадратного корня из предыдущей величины, и это даёт
qr(1) = (8pIkT)1//h.
Было бы правильно различать внешние и внутренние вращательные степени свободы.
Обычно система уровней вращения молекулы определяются геометрической конфигурацией её ядерного остова. Существует несколько моделей такого распределения. Их называют волчками разных типов.
На основе квантовой механики для волчков различных типов невозможно получить общее аналитическое выражение ротационных сумм состояния.
Тем не менее, для этой цели существуют достаточно точные приближённые приёмы, основанные на классических способах описания вращения достаточно тяжёлых молекул с близко расположенными ротационными уровнями.
3) Коррекция трансляционной суммы состояний состоит в учёте неразличимости частиц относительно перестановки – существует перестановочная симметрия коллектива неразличимых частиц. Свойства коллектива не изменяются при обмене одинаковых частиц местами в пространстве. Таких вариантов обмена столько же, сколько перестановок в этом коллективе.
Поэтому необходимо перейти к пространственному движению коллектива, и это даст:
qt-loc(3) ≈ [qt(1)]3 = [(2pmkT)1/L/h]3 ® q t-loc(3) =[(2pmkT)3//h3]V
qt-loc (3N)≈[q t- loc (3)]N =[(2pmkT)3/V/h3]N ¬(PN=N!)® qt-deloc(3N)= qt-loc(3N)/N!®
®qt-deloc(3N)=[(2pmkT)1/V/h]3N/N! ºqt(3N)®
Необходимо заменить факториал.
Математическая справка для замены:
Факториал больших чисел - формула Стирлинга:
N!@(2pN) 1/2(N/e)N ≈(N/e)N
Для вычисления логарифмов огромных чисел (при переходе от термодинамических вероятностей к энтропии) это приближение очень хорошее.
Заменяя факториал, получаем более простое выражение:
® qt(3N) ={[(2pmkT)1/V/h]3}Ne N/N N={[(2pmkT) 3/2/h3]eV/N}N;®
qt(3) =[(2pmkT) 3/2/h3](e/N)V.
Поступательная сумма, отнесённая к одной частице
qt(3) =[(2pkT)3/2/h3](e/N)[m3/2V]
Молекулярная сумма состояний.
Qtrven,i=qtiqei(Piqri)(Piqvi(Pqni);
Qi=(Qtrven,i)N;
Свободная энергия трансляционного движения.
Ati=-kT×lnQi=-NkT×lnQtrven,i;
Энергия Гиббса трансляционного движения.
Gti =Ai+piV=Ai+NikT;
Химический потенциал трансляционного движения.
mi=Gi/Ni;
Трансляция, сумма состояний, вклад в свободную энергию, давление газа.
С нею непосредственно связано давление газа, поскольку только в ней представлен объём.
Qt = [(2pmkT)3/2/h3]e(V/N) ; (на одну частицу)
At =NkTlnQt; (на коллектив частиц)
dA= -pdV-SdT;
p= -(¶A/¶V)T = NkT(¶lnQt/¶V)T;
ln Qt = ln[(2pmkT)3/2/h3]+1+lnV-lnN;
ln Qt = ln [(2pmk)3/2/h3]+ (3/2)lnT+1+lnV-lnN;
(¶lnQt/¶V)T =1/V;
p= NkT(1/V);
At = -NkT×ln[(2pmkT)3/2/h3]-NkT-NkT×ln(V/N);
pV= NkT; ®Vo= NkT/Po.
Прочие степени свободы движения объёма не содержат и на давление не влияют.
...Но влияют на функции состояния.
G = A+PV = -NkT×ln[(2pmkT)3/2/h3]-NkT-NkT×lnV+NkT×lnN+NkT; ®
G = -NkT×ln[(2pmkT)3/2/h3]-NkT× ln(V/N) ; ®
G = -NkT×ln[(2pmkT)3/2(V/N)/h3] ; ®
G ot = -NkT×ln[(2pmkT)3/2(Vo/N)/h3] ; ®
Got = -NkT×ln[(2pmkT)3/2(kT/Po)/h3].
Мольный изобарный потенциал это химический потенциал:
Got ºm ot®
mot =-NAkT×ln[(2pmkT)3/2(kT/Po)/h3] ;
В химическом потенциале представлены все виды движения:
moi=moti+mori +moVi +moei +moni ;
moi = -NAkT×ln[Qio] ;
В обычном термодинамическом смысле стандартизация по давлению затрагивает только поступательную сумму состоянийю.
moi=-NAkT×{ln[(2pmikT)3/2(kT/Po)/h3]+ln[Qri]+ln[QViQeiQni] };
Вырожденности электронного и ядерного термов молекул-участников реакции называют также статистическими весами.
Доступные в химии дистанции между ядерными уровнями различаются очень мало, и ядерные факторы Больцмана почти не различимы, и ядерные суммы состояний это просто множители – вырожденности:
Qni=gni=2Ini+1.
Электронные суммы состояний содержат почти всегда лишь по одному фактору Больцмана,
а в качестве сомножителей и электронные вырожденности, так что это полное число входящих в него микросостояний, домноженное на фактор Больцмана с электронным термом:
Qei = gei´exp(-Ei min/kT)
Результат в виде стандартного химического сродства (приращения энергии Гиббса за пробег реакции) равен:
DrGo = -RTln Kp=-NAkT ln Kp=Sni×moi , откуда получается выражение логарифма константы равновесия. Это компактная форма

Потенцирование этого выражения даёт простейший результат:

Любая термодинамическая функция состояния может быть получена из знакомых термодинамических потенциалов. В наших расчётах удобна свободная энергия:
A = -NkT×lnQ[(m, I1, I2, … n1,n 2, …)( T,V)].
p = -(¶A/¶V)T = NkT;
A ® A+pV = G ®m; Ao+NkT
¯
S(T,V)= - (¶A/¶T)V = - (¶G/¶T)p ;
¯
U(T) = A+TS ® CV = (¶U/¶T)V;
¯
H(T) =U+pV=U+NkT ® Cp = (¶H/¶T)V = CV + Nk .
Выполненные расчёты демонстрируют удивительные возможности, предоставляемые статистической термодинамикой. Формальная термодинамика таких возможностей не даёт...
|