Курсовая работа: Определение металлических примесей методом атомно-абсорбционной спектрометрии в марганце марки
|
Название: Определение металлических примесей методом атомно-абсорбционной спектрометрии в марганце марки Раздел: Рефераты по химии Тип: курсовая работа | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
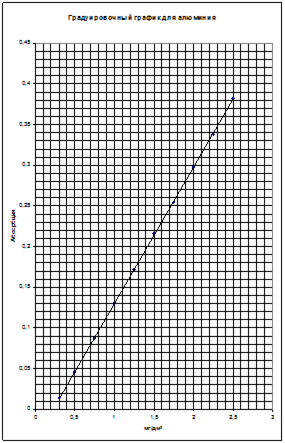
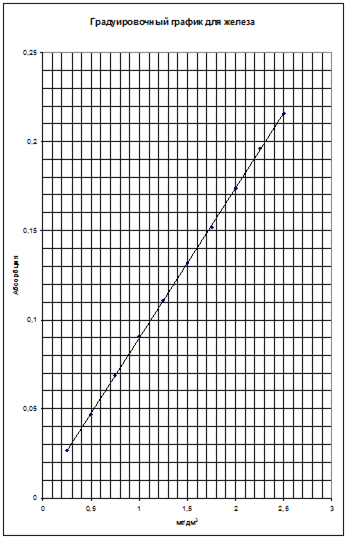
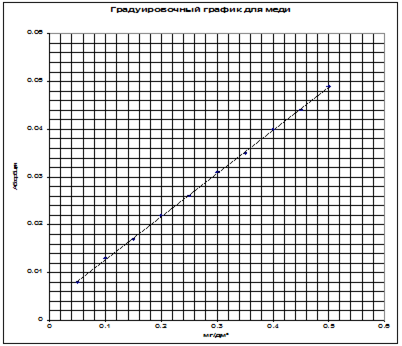
| Федеральное агентство по образованию РФ Государственное образовательное учреждение Высшего профессионального образования Тульский государственный университет Кафедра химии Курсовая работа по аналитической химии Определение примесей алюминия, железа, меди, никеля, титана, кальция и магния методом атомно-абсорбционной спектрометрии в образцах марганца марки Мн 998. Выполнила: студентка гр. 430481 Родичева А.С. Научный руководитель: д. х. н., доцент Егоров А.М. Тула, 2010 СОДЕРЖАНИЕ ВВЕДЕНИЕ 1. ЛИТЕРАТУРНЫЙ ОБЗОР 1.1 История появления метода 1.2 Характеристика атомно-абсорбционного анализа 1.2.1 Общая характеристика метода 1.2.2 Схемы установок атомно-абсорбционных спектрометров 1.2.3 Источники излучения 1.2.3.1 Естественное уширение 1.2.3.2 Допплеровское уширение 1.2.3.3 Лоренцевское уширение 1.2.3.3 Лампы с полым катодом 1.2.4 Атомизаторы 1.2.4.1 Пламенный атомизатор 1.2.4.1.1 Процессы, происходящие в пламени 1.2.4.2 Электротермический атомизатор 1.2.4.3 Гидридная техника 1.2.5 Монохроматоры 1.2.6 Детекторы 1.2.7 Количественный анализ 1.2.8 Факторы, влияющие на величину абсорбционного сигнала 1.2.8.1 Коррекция фонового поглощения 1.2.8.1.1 Применение дейтериевой лампы 1.2.8.1.2 Метод двух линий 1.2.8.1.3 Применение эффекта Зеемана 1.2.9 Помехи и способы их устранения при проведении атомно-абсорбционного анализа 1.2.9.1 Влияния в пламени 1.2.9.2 Методы устранения мешающих влияний в пламенном атомно-абсорбционном анализе 1.2.9.3 Влияния в графитовой печи 1.2.9.4 Способы устранения мешающих влияний 1.2.10 Метрологические характеристики метода 1.2.11 Область применения атомно-абсорбционной спектрометрии 1.3 Определение примесей в марганце различными методами анализа 1.3.1 Фотометрическое определение фосфора в металлическом и азотированном марганце 1.3.1.1 Фотометрический метод с применением аскорбиновой кислоты 1.3.1.2 Фотометрический метод с применением тиомочевины или ионов двухвалентного железа 1.3.2 Фотометрическое и гравиометрическое определение кремния в металлическом и азотированном марганце 1.3.2.1 Фотометрическое определение кремния в металлическом и азотированном марганце 1.3.2.2 Гравиметрический метод кремния в металлическом и азотированном марганце 1.3.3 Фотометрическое, атомно-абсорбционное и титриметрическое определения железа в металлическом и азотированном марганце 1.3.3.1 Фотометрическое определение железа в металлическом и азотированном марганце с применением 1,10-фенантроина 1.3.3.2 Фотометрический метод с применением сульфосалициловой кислоты 1.3.3.3 Атомно-абсорбционный метод 1.3.3.4 Титриметрический метод 1.3.4 Фотометрические и атомно-абсорбционные методы определения никеля в металлическом марганце и металлическом азотированном марганце 1.3.4.1 Фотометрическое определение никеля в металлическом марганце и металлическом азотированном марганце 1.3.4.2 Атомно-абсорбционное определение никеля в металлическом марганце и металлическом азотированном марганце 1.3.5 Фотометрическое и атомно-абсорбционное методы определения меди в металлическом марганце и металлическом азотированном марганце 1.3.5.1 Фотометрическое определение меди в металлическом марганце и металлическом азотированном марганце 1.3.5.2 Атомно-абсорбционное определение меди в металлическом марганце и металлическом азотированном марганце 1.3.6 Атомно-абсорбционное методы определения кальция и магния металлическом марганце и металлическом азотированном марганце 1.3.7 Фотометрический метод определения алюминия в металлическом марганце и металлическом азотированном марганце 1.3.8 Фотометрический метод определения титана в металлическом марганце и металлическом азотированном марганце 1.3.9 Определение марганца в металлическом и азотированном марганце 2. ЭКСПЕРЕМЕНТАЛЬНАЯ ЧАСТЬ 2.1 Оборудование и реактивы 2.2 Подготовка к проведению анализа и приготовление серии стандартных растворов 2.3 Отбор и хранение проб 2.4 Приготовление исходных растворов марганца 2.5 Определение систематической ошибки приготовления растворов марганца 2.6 Методика определения металлических примесей в образцах марганца марки Мн 998 методом атомно-абсорбционной спектрометрии согласно ГОСТ 16698.6-71, ГОСТ 16698.7-71, ГОСТ 16698.9-71, ГОСТ 16698.10-71 2.7 Формулы для статистической обработки полученных результатов 3. ОБРАБОТКА РЕЗУЛЬТАТОВ 3.1 Качественное определение металлических примесей в марганце марки Мн 998 3.2 Построение градуировочного графика для алюминия 3.3 Статистическая обработка графика градуировочной зависимости для алюминия 3.4 Построение градуировочного графика для железа 3.5 Статистическая обработка графика градуировочной зависимости для железа 3.6 Построение градуировочной зависимости для меди 3.7 Статистическая обработка графика градуировочной зависимости для меди 3.8 Построение градуировочной зависимости для никеля 3.9 Статистическая обработка графика градуировочной зависимости для никеля 3.10 Количественное определение алюминия в образцах марганца марки Мн 998 3.11 Количественное определение железа в образцах марганца марки Мн 998 3.12 Количественное определение меди в образцах марганца марки Мн 998 3.13 Количественное определение никеля в образцах марганца марки Мн 998 3.14 Количественная характеристика примесей алюминия, железа, меди, никеля в образцах марганца марки Мн 998 3.15 Значения относительных стандартных отклонений, коэффициентов чувствительности и нижних границ определяемых концентраций для металлических примесей обнаруженных в составе марганца марки Мн 998 ВЫВОДЫ СПИСОК ЛИТЕРАТУРЫ ВВЕДЕНИЕ
Метод атомно-абсорбционной спектрометрии является универсальным количественным методом для определения небольших количеств элементов (порядка 10-4 или 10-5 %) в большинстве природных (почвах, удобрениях, растениях, пищевых продуктах, нефти, смазочных маслах, питьевых, природных и сточных водах, морской воде, воздухе, и т. д.) и технических (металлы, сплавы, продукты гидрометаллургической переработки руд и т. д.) объектах. Данным методом можно определить почти 80 элементов в их числе Al, Mg, Сu, Pb, Fe, Ag, Ni, Hg, Cd, Cr, Mn. Метод обладает высокой чувствительностью (для большинства элементов составляют 10-6 - 10-4 в пламенном и 10-9 - 10-7 % масс) и селективностью, широким диапазоном определяемых концентраций (диапазон значений обычно составляет от нескольких сотых до 0,6 - 1,2 единиц оптической плотности), и воспроизводимостью. Целью данной работы является качественное и количественное определение методом атомно-абсорбционной спектрометрии примесей алюминия, железа, меди, никеля, титана, кальция и магния в образцах марганца марки Мн 998. Задачами настоящей работы является: 1. освоение методик атомно-абсорбционной спектрометрии 2. качественное и количественное определение металлических примесей в марганце марки Мн 998 3. освоение методов статистической обработки данных. 1. ЛИТЕРАТУРНЫЙ ОБЗОР
1.1 История появления метода
Открытие и история исследований атомной абсорбции связано со всей историей спектрального анализа. В 1802 году У. Волластон наблюдал темные линии в спектре солнца. В 1814 году Й. Фраунгофер определил длины волн этих линий. Р. Бунзен раскрыл причину их возникновения в солнечном спектре: атомы каждого элемента поглощают свет той же длины волны, что и испускают. В 1861 году Г. Кирхгоф определил законы такого поглощения (атомной абсорбции) и установил линейную зависимость между величиной поглощения света и концентрацией поглощаемых атомов. Датой рождения нового метода можно считать 1861 год, когда была опубликована статья Г. Кирхгофа по спектральному анализу химического состава солнечной атмосферы [1]. В аналитической химии атомно-абсорбционный метод долго не находил применения так как отсутствовал подходящий источник света. Единственным подходящим источником была ртутная лампа, на основе которой Вудсон создал первый в мире атомно-абсорбционный спектрометр и запатентовал метод определения ртути в воздухе, воспользовавшись тем фактом, что ее пары находятся в атомном состоянии даже при комнатной температуре. Широкое применение в химии данный метод получил после работы А. Уолша в 1955 году, в которой он применил в качестве источника излучения лампу с полым катодом и пламя в качестве атомизатора [1]. Общие требования, предъявляемые к атомно-абсорбционной аппаратуре, сформулированные Уолшем, сводятся к следующему: для проведения анализа необходимо располагать источником соответствующего излучения, средством получения атомного пара определяемого элемента, прибором для выделения абсорбционного сигнала, средством для его регистрации и измерения и стандартами, с которыми должен сравниваться анализируемый образец. Из выше сказанного можно заметить, что атомно-абсорбционный метод не уступает по своей простоте эмиссионному пламенно-фотометрическому методу, являющемуся, как известно, наиболее простым и наиболее доступным методом спектрального анализа [1]. Значительный вклад в развитие метода внесли русские ученые. Б. В. Львов предложил конструкцию графитовой кюветы для непламенного атомизатора; Н. С. Полуэктов с сотрудниками разработал способ беспламенного определения ртути в воде, донных осадках и биологических объектах; И. А. Нивчук – методику определения летучих элементов в стали и алюминиевых сплавах; К. П. Столяров и А. К. Чарыков предложили методы экстракционного атомно-абсорбционного определения элементов в объектах окружающей среды [1]. В настоящее время работы по атомно-абсорбционной спектрометрии ведутся в различных направлениях: разрабатываются методы анализа газов и изотопного состава элементов; ведутся измерения абсолютных величин сил осцилляторов и ширины резонансных линий, коэффициентов диффузии паров элементов в инертных газах. Особенно широко метод используется для анализа элементного состава вещества [1]. 1.2 Характеристика атомно-абсорбционного анализа 1.2.1 Общая характеристика метода Ι. Атомно-абсорбционный анализ – один из наиболее чувствительных, быстрых, точных и селективных методов современной аналитической химии. Основным его достоинством является селективность. Возможность взаимного наложения резонансных линий различных элементов при атомно-абсорбционных измерениях практически исключена. Из одного раствора можно определить большое количество элементов без разделения [2]. Этим метод атомной абсорбции выгодно отличается от эмиссионного спектрального анализа и молекулярной спектрометрии, которые вследствие большого количества спектральных помех требует определения или маскирования мешающих элементов или использование градуировочных образцов, идентичных по матричному составу анализируемым пробам [2]. Кроме того, в эмиссионном анализе регистрируется излучение возбужденных атомов, концентрация которых сильно зависит от температуры, и даже небольшое ее изменение влияет на интенсивность аналитического сигнала [2] . Для абсорбционного же анализа существенно количество атомов, находящихся в возбужденном состоянии. Благодаря этому обстоятельству значительно уменьшается взаимное влияние компонентов образца, что дает возможность использовать для градуировки в большинстве случаев водные растворы определяемого элемента [2]. ΙΙ. Атомы вещества в парообразном состоянии при определенных условиях способны излучать лучи определенных длин волн (так называемое характеристическое излучение), причем для каждого элемента она своя. Вместе с тем атомы способны поглощать то излучение, которое сами испускают. Таким образом, длина волны лучей, поглощенных атомами элемента, совпадает с длиной волны его характеристических излучений [3]. Методика атомно-абсорбционного спектрального анализа заключается в том, что исследуемое вещество вводят в газовое пламя (для пламенного атомизатора), одновременно пламя освещают светом с непрерывным спектром, например от лампы накаливания или от трубки с полым катодом (газоразрядная трубка, в спектре которой наблюдаются линии элементов, входящие в состав материала катода). В полученном спектре интенсивность света в области характеристических частот будет меньше интенсивности ближайших соседних участков спектра. Ослабление интенсивности в области характеристических частот измеряют при помощи фотоэлектрической установки. Между ослаблением интенсивности линии, характерной для данного элемента, и концентрацией этого элемента в исследуемой пробе наблюдается линейная зависимость [3]. Результат анализа в атомно-абсорбционной спектрометрии зависит главным образом от числа невозбужденных атомов, которое в известных пределах сравнительно мало изменяется с температурой. Это уменьшает эффекты взаимного влияния компонентов пробы на аналитический сигнал. В атомно-абсорбционной спектрометрии практически полностью исключена возможность наложения линий различных элементов, так как в условиях атомно-абсорбционного анализа число линий в спектре значительно меньше, чем в эмиссионной спектроскопии [4]. Методом атомно-абсорбционной спектрометрии можно определять почти 90 элементов, главным образом металлов. Неметаллы, как правило, непосредственно определять нельзя. В то же время существуют способы косвенного определения неметаллов по величине поглощения молекулярных полос. Атомно-абсорбционный метод широко используют как метод массовых, быстрых, селективных и достаточно точных определений металлов. Методом атомно-абсорбционной спектрометрии принципиально возможно определять как следовые, так и достаточно высокие содержания (в последнем случае — после соответствующего разбавления). Чаще всего этим методом определяют малые содержания: в пламенной атомно-абсорбционной спектрометрии — порядка нанограммов-микрограммов на миллилитр, в электротермической — пикограммов-нанограммов на миллилитр. В электрическом атомно-абсорбционном спектрометре можно определить элементы, концентрация которых в пробе составляет фемтограммы, объем самой пробы при этом всего 10-200 мкл [1]. Недостаток атомно-абсорбционной спектрометрии состоит в том, что это одноэлементный метод анализа. Для определения каждого элемента необходимо использовать свою лампу с полым катодом. Для достаточно быстрого определения нескольких элементов можно установить несколько ламп во вращающийся барабан и поочередно облучать атомизатор. Однако производительность такого устройства все же недостаточно высока, а соотношение “производительность — затраты” ниже, чем для атомно-эмиссионного метода. Трудности могут возникнуть и при определении методом атомно-абсорбционной спектрометрии с электротермической атомизацией сверхмалых количеств элементов в матрицах сложного состава. В подобных случаях для получения правильных результатов необходимо сочетание атомно-абсорбционной спектрометрии с химическими методами пробоподготовки, например, отделения определяемого компонента от матрицы с помощью ионообменной хроматографии [5]. 1.2.2 Схемы установок атомно-абсорбционных спектрометров Существует два вида атомно-абсорбционные спектрометров: пламенный и электротермичекий, (основное различие между ними заключается в структуре атомизатора). Схема спектрометра, использующего пламенный атомизатор, представлена на рис. 1. Такой спектрометр состоит из источника излучения (1), атомизатора – пламени (2), монохроматора (3) и детектора – приемника света (4), и двухлинзовой оптической системы (5).
Рис. 1. Спектрометр с электротермичеким атомизатором (схема представлена на рис. 2) состоит из источника излучения (1), оптической системы линз (2), электротермического атомизатора, включающего графитовую трубчатую печь (3) и электромагнит (4), монохроматора (5), фотоэлектрического преобразователи с подключенным к нему персональным компьютером (6) [1].
Рис. 2. Проба вносится в атомизатор, где распадается до свободных атомов. Возбуждение атомов осуществляется потоком света УФ-видимой области, исходящего из лампы с полым катодом. Отсечение постороннего излучения и детектирование производятся при помощи — монохроматора и фотоэлектронного умножителя (ФЭУ), соединенного с устройством отображения информации [5]. 1.2.3 Источники излучения Чтобы измерить степень поглощения света анализируемым образцом, необходимо сравнить интенсивности света, падающего на образец и прошедшего через образец [5]. Спектры молекул имеют достаточно широкие полосы поглощения. В молекулярной спектроскопии используют источники излучения, дающие непрерывный спектр. Из него с помощью монохроматора выделяют спектральную полосу, лежащую в требуемом диапазоне [5]. В атомно-абсорбционной спектрометрии применение источников непрерывного спектра невозможно. Причина состоит в том, что атомные линии поглощения очень узкие, их ширина составляет 10-3 - 10-2 нм. При облучении атомов недостаточно монохроматичным источником света большая часть светового потока пройдет через образец без поглощения [5]. Поэтому в атомно-абсорбционной спектрометрии необходимо использовать источники света, дающие линейчатый спектр. При этом ширина линий в спектре испускаемого света должна быть, по крайней мере, сравнима с шириной линий атомного спектра [5]. Ширина атомных спектральных линий зависит от многих факторов. Естественное уширение, дополнительное уширение вызвано эффектом Допплера. Ширина линий зависит также от давления в атомизаторе и интенсивностей электрического и магнитного полей [5].
1.2.3.1 Естественное уширение Естественное уширение. А. Уолш указал, что естественная ширина спектральной линии имеет порядок 10-5 нм. Она зависит от степени расширения уровней, определяемой временем пребывания электрона на верхнем энергетическом уровне. Естественным уширением можно пренебречь, так как оно незначительно [5].
1.2.3.2 Допплеровское уширение Эффект Допплера заключается в изменении частоты излучения при движении излучателя и приемника излучения друг относительно друга. В акустике этот эффект можно наблюдать в повседневной жизни: когда мимо вас на большой скорости проносится автомобиль, издающий громкий звуковой сигнал, высота звука непрерывно изменяется. В атомно-абсорбционной спектроскопии излучающими объектами являются атомы, совершающие беспорядочное тепловое движение в атомизаторе в разных направлениях относительно неподвижного приемника излучения. Их скорости движения подчиняются закону распределения Максвелла-Больцмана. Средняя скорость движения атомов пропорциональна корню квадратному из температуры. Атомы, движущиеся в направлении распространения излучения, поглощают при более низких частотах, а движущиеся навстречу излучению — при более высоких. В результате возникает симметричное уширение спектральной линии, называемое допплеровским. Его величина приблизительно в 100 раз больше, чем естественного. Уравнение (1) описывает зависимость допплеровского уширения от температуры (Т), длины волны излучения (λ) и массы атома (М): ∆λ = где k– постоянная Больцмана, c - скорость света [5]. 1.2.3.3 Лоренцевское уширение Еще одной причиной уширения линий являются столкновения атомов в атомизаторе с другими атомами или ионами. Их вероятность повышается с ростом давления. Вследствие взаимодействия электронных состояний сталкивающихся частиц наблюдается расщепление их энергетических уровней и, следовательно, уширение спектральных линий. Этот тип уширения называется лоренцевским. Величина лоренцевского уширения также на два-три порядка выше, чем естественного. В пространстве лампы с полым катодом излучающие атомы находятся под давлением ниже атмосферного. Поэтому линии спектра испускания лампы более узкие, чем линии спектра поглощения атомов в атомизаторе [5]. 1.2.3.4 Лампы с полым катодом Таким образом, в атомно-абсорбционной спектрометрии необходимо использовать источники излучения с шириной линий менее 10-3 - 10-2 нм. В качестве таковых обычно используют лампы с полым катодом (ЛПК), с определенным элементом. Излучение этих ламп обусловлено процессами возбуждения атомов при низкой температуре — более низкой, чем температура атомизатора. По этой причине ширина линий спектра лампы также меньше, чем ширина атомных линий поглощения. Столь монохроматичное излучение в заметной мере поглощается атомами. Посторонние линии, присутствующие в спектре ЛПК, отрезаются монохроматором [5]. Строение лампы с полым катодом изображено на рис. 3. Сама лампа представляет собой цилиндрический стеклянный баллон с кварцевым или стеклянным окошком, заполненным аргоном или неоном при пониженном давлении (200 - 800 Па), в котором происходит испарение вещества и возбуждение атомов элемента при электрическом заряде в атмосфере инертного газа. Лампа с полым катодом испускает интенсивные узкие линии элемента, входящего в состав катода.
Рис. 3. Схема строения лампы с полым катодом [5]. Анод такой лампы – металлическая вольфрамовая проволока, находящаяся рядом с катодом. Катод представляет собой полый цилиндр, изготовленный из определенного элемента или его сплава. Катод и анод размещены в стеклянном цилиндре. Когда на электроды лампа поступает напряжение от высокоточного выпрямителя 600 В, газ ионизируется. Катионы газа выбирают из катода атомы определенного элемента и возбуждают их термически. При обратном переходе возбужденных атомов в основное состояние излучается свет определенных длин волн. В спектре свечения при температуре 800 К в полом катоде наблюдаются резонансные частоты элементов. Ширина линий испускания ЛПК составляет 10-4 - 10-3 нм [1]. Металл, используемый для изготовления ЛПК, должен быть высокой чистоты и не содержать адсорбционный водород. Работа лампы ухудшается из-за снижения давления газа вследствие частичной его сорбции на катоде. Из некоторых материалов — As, Sb, Se, Те — трудно изготовить полый катод. Кроме того, излучение, даваемое этими неметаллами, часто находится в весьма коротковолновой области. Для его возбуждения требуется значительная энергия, а его интенсивность низка. В подобных случаях вместо ламп с полым катодом применяют безэлектродные разрядные лампы. Внутри такой лампы находится небольшое (1 - 2 мг) количество соответствующего элемента, который во время работы лампы под действием высокочастотного электрического разряда переходит в парообразное состояние. Как и для ЛПК, корпус безэлектродной разрядной лампы сделан из кварца, а внутреннее пространство заполнено инертным газом при пониженном давлении [1]. Серьезный недостаток разрядных ламп – их “узкая специализация”: каждая лампа пригодна для определения только одного элемента. Существуют и многокомпонентные лампы, в которых катод изготовлен из смеси (сплава) нескольких элементов, но у них эксплуатационные характеристики, как правило, хуже, чем у одноэлементных. Поэтому предпринимаются усилия по созданию источников излучения для атомно-абсорбционной спектрометрии с перестраиваемой частотой. Примеры таких источников – особо мощные (ксеноновые) лампы, дающие непрерывный спектр, в сочетании с монохроматорами с высокой разрешающей способностью, а также лазеры с перестраиваемой частотой: на красителях и, в последнее время, - на полупроводниковых диодах. Излучение последних отличается столь высокой монохроматичностью, что позволяет определять даже изотопы элементов, используя очень малые различия в положении их спектральных линий. Тем не менее, лампы с полым катодом и безэлектродные разрядные лампы до сих пор используются в атомно-абсорбционной спектрометрии наиболее широко [5]. 1.2.4 Атомизаторы Атомизатор – это устройство, используемое для переведения определяемого элемента в атомный пар с возможно большей эффективностью. В атомно-абсорбционном анализе атомизация элемента достигается нагреванием пробы до 2000 – 3000 о С. Простейшим способом перевода растворенной пробы в атомарное состояние является использование пламени (пламя горючих газов в смеси с окислителями). К самому пламени предъявляются следующие требования: · оно должно быть прозрачным (т. е. иметь высокую пропускаемость) во всем спектральном диапазоне – от 193 до 852 нм.; · собственное излучение пламени должно быть настолько слабым, чтобы модулятор позволял устранить влияние этого излучения; · эффективность атомизации элемента в пламени должна быть как можно больше; · степень ионизации элемента в пламени должна быть незначительна. Эти требования иногда противоречат друг другу. Например, высокотемпературное пламя обеспечивает высокую степень атомизации, но и приводит к увеличению ионизации определяемого элемента [2]. Впоследствии для улучшения чувствительности определения был предложен электротермический способ атомизации с использованием графитовых печей. Рассмотрим сначала пламенные атомизаторы [1]
1.2.4.1 Пламенный атомизатор При пламенном способе атомизации раствор пробы распыляют в пламя в виде мелких капель. Устройство атомизатора этого типа на основе щелевой горелки, дающей ламинарное пламя, изображено на рис. 4. Продольная длина пламени составляет 5 - 10 см.
Рис. 4. Схема атомизатора для пламенной атомно-абсорбционной спектрометрии на основе щелевой горелки. I0 – интенсивность падающего света, I – интенсивность прошедшего света. Горючая смесь для поддержания пламени состоит из горючего газа и газа-окислителя. Окислитель может одновременно служить распыляющим газом или подаваться в горелку отдельно (вспомогательный газ). Наиболее распространенные сочетания газов приведены в таблице №1. Для определения большинства элементов достаточно температур, даваемых смесью ацетилен-воздух. Это пламя имеет наибольшее применение в атомно- –абсорбционном анализе. Оно наиболее стабильно, его стехиометрию можно регулировать в широких пределах – от сильно окислительного (с большим избытком воздуха) до сильно восстановительного (с избытком ацетилена). В отличие от других пламен температура пламени ацетилен – воздух почти не зависит от высоты и стехиометрии [6]. Таблица №1. Составы газовых смесей для пламенной атомно-абсорбционной спектрометрии.
Недостаток ацетилен-воздушного пламени — значительное собственное поглощение при длинах волн менее 230 нм. Для трудно атомизируемых или труднолетучих элементов — Be, Ca, Sr, Ti, Zr, Hf, V, Mo, W, редкоземельные элементы — необходимо применение более высоких температур. Для этого можно использовать смесь ацетилен-закись азота, дающую температуру до 3100 К. Однако столь горячее пламя обладает большим собственным испусканием. Для элементов, атомы которых поглощают в коротковолновой области (As, Se), целесообразно использовать пламя водород-воздух, не обладающее заметным поглощением вплоть до 210 нм. Для определения легко атомизируемых щелочных металлов достаточно температуры пламени метан-воздух [1]. 1.2.4.1.1 Процессы, происходящие в пламени В пламени происходит испарение составных частей пробы, их диссоциация на свободные атомы, возбуждение атомов под действием внешнего излучения и, как побочный процесс, ионизация атомов. Эти же процессы протекают и в атомизаторах других типов, рассматриваемых далее [5]. Испарение. Первым компонентом пробы, переходящим в газообразное состояние, является растворитель. Затем испаряются твердые компоненты, находящиеся в растворе. При использовании органических растворителей наблюдается их горение. Испарение твердых компонентов может происходить непосредственно или через стадию плавления. Последний случай сопровождается гомогенизацией пробы и может привести к образованию весьма труднолетучих смешанных оксидов или фосфатов. В этих случаях необходимо правильно подобрать состав и температуру пламени: температура должна быть достаточно высокой, а среда в пламени — восстановительной. Подобными свойствами обладает, например, пламя ацетилен-закись азота, применяемое для определения Аl, В, Si [5]. Мешающее влияние матрицы можно устранить также с помощью добавок специальных реагентов. Так, при определении кальция в фосфатных растворах добавляют хлорид лантана в качестве “освобождающей” добавки. В пламени лантан образует фосфат LaPO4 , предотвращая тем самым образование труднолетучего пирофосфата кальция. Можно использовать и добавку ЭДТА. Кальций при этом образует комплекс с ЭДТА, который более устойчив, чем фосфатный, и легко атомизируется вследствие сгорания органического лиганда [5]. Диссоциация и восстановление. На следующей стадии испарившиеся соединения металлов диссоциируют на свободные атомы. Процесс диссоциации сопровождается одновременным восстановлением ионов до свободных атомов металлов:
Подобные равновесия можно охарактеризовать с помощью обычных величин, применяемых для описания химических равновесий — степени диссоциации α и константы диссоциации KD . Константу в этом случае выражают через парциальные давления компонентов: KD
= α = Степень диссоциации зависит от температуры пламени, энергии диссоциации соединения, его концентрации и степени влияния на положение равновесия со стороны посторонних компонентов [5]. С увеличением температуры степень диссоциации возрастает, поскольку при этом увеличивается константа диссоциации KD . Энергии диссоциации некоторых молекул приведены в таблице №2. Ввиду различий в энергиях диссоциации молекул степень диссоциации может зависеть от валового состава пробы. Наличие посторонних веществ может сказываться на величине аналитического сигнала и вследствие их влияния на положение равновесия диссоциации. Например, в присутствии высоких содержаний КСl равновесие диссоциации хлорида натрия (2) смещается влево (из-за возрастания pCl ), и степень диссоциации снижается. Поэтому градуировочные зависимости, построенные с использованием чистых водных растворов NaCl, могут оказаться непригодными для определения натрия в присутствии КС1. Чтобы избежать погрешностей, следует строить градуировочные зависимости, используя растворы, близкие по составу к анализируемой пробе [5]. Из уравнения (4) следует также, что степень диссоциации уменьшается с увеличением концентрации определяемого вещества. Вследствие этого может нарушиться пропорциональная зависимость между концентрацией натрия в анализируемом растворе и парциальным давлением атомов натрия в пламени и, как результат, наблюдаться искривление градуировочной зависимости [5]. Таблица №2. Энергии диссоциации некоторых молекул.
Возбуждение. Число невозбужденных свободных атомов, способных к возбуждению под действием излучения лампы с полым катодом, определяется в соответствии с распределения Больцмана:
где N* - число частиц в возбужденном состоянии, N0 - число частиц в основном состоянии при тепловом равновесии; g* , g0 – статистические веса возбужденного и основного состояний; ∆E – разность энергий возбужденного и основного состояний; k – константа Больцмана (1,38·10-23 Дж·К-1 ). Ионизация. Наряду с диссоциацией происходит (особенно интенсивно — при высоких температурах) и нежелательный процесс ионизации свободных атомов:
Для этого равновесия можно записать выражения константы ионизации (KI ) и степени ионизации (β): KI
= β = Чем ниже энергия ионизации, тем выше доля ионизированных атомов. Как видно из таблицы №3, явление ионизации особенно заметно в случае щелочных металлов. Таблица №3. Энергии ионизации атомов некоторых металлов.
Положение равновесия ионизации непосредственно зависит от парциального давления свободных электронов в пламени. Оно, в свою очередь, может зависеть от состава матрицы. Для поддержания парциального давления электронов на постоянном и высоком уровне к пробе часто (особенно при использовании высокотемпературных пламен таких, как ацетилен-закись азота) добавляют избыток соли легко ионизирующегося элемента, например, натрия или калия. Такие добавки называют спектроскопическими буферами [5]. 1.2.4.2 Электротермический атомизатор Еще один способ атомизации состоит в использовании графитовых трубок, нагреваемых электрическим током. Их часто называют графитовыми кюветами (Львов, 1958 год, Массман, 1970 год). На рис. 5 показано устройство такой графитовой кюветы. Длина трубки составляет обычно от 30 до 50 мм, внутренний диаметр — около 10 мм. Раствор пробы (порядка 10 мкл) вводят в кювету и нагревают ее по специальной температурной программе, подводя напряжение через металлические контакты. Таким способом можно достичь температур порядка 3000 К [5].
Рис. 5. Схема графитовой кюветы для электротермическрй атомизации. I0 – интенсивность падающего света, I – интенсивность прошедшего света. Путем программируемого повышения температуры до 105-110°С раствор пробы сначала высушивают в защитной атмосфере инертного газа (например, аргона). Затем пробу озоляют, поднимая температуру до 500 — 700°С. В процессе озоления удаляются летучие компоненты матрицы — соединения ртути, органические вещества, некоторые галогениды. При этом также протекает ряд реакций разложения — дегидратация кристаллогидратов и гидроксидов, разложение нитратов. Кроме того, многие компоненты пробы под действием графита восстанавливаются: сульфаты до сульфидов, некоторые ионы металлов — до свободных металлов. При этом также могут образоваться и нежелательные побочные продукты: термически устойчивые карбиды или труднолетучие оксиды, например, бора или фосфора. Затем температуру повышают до 2000-3000 К. При этом происходят процессы диссоциации, восстановления и ионизации, аналогичные описанным ранее в применении к пламенным атомизаторам [5]. 1.2.4.3 Гидридная техника Очень эффективный способ атомизации состоит в превращении определяемого компонента в летучее соединение и вводе его в пламенный (обычно используют водородно-воздушное пламя) или графитовый атомизатор в виде пара или газа. Таким образом можно определять ртуть, обладающую значительным давлением насыщенного пара уже при обычных условиях. Такие элементы, как As, Bi, Ge, Sb, Se, Sn, переводят в летучие гидриды восстановлением боргидридом натрия NaBH4 . Сравнение пламенного и электротермического способов атомизации [5]. В целом о возможностях двух описанных способов атомизации в атомно-абсорбционной спектрометрии можно сказать следующее: Чувствительность. При электротермическом способе атомизации в атомизатор попадает все количество пробы, а при распылении в пламя — не более 10%. Время пребывания пробы в электротермическом атомизаторе значительно выше, чем в пламенном. Вследствие этого пределы обнаружения при использовании электротермических атомизаторов обычно на несколько порядков ниже [5]. Для повышения эффективности атомизации в пламени в последнее время применяют способ прямого ввода раствора пробы в атомизатор (пламенно-инжекционная техника) [5]. Селективность. Электротермический способ атомизации позволяет непосредственно в ходе анализа удалить из пробы часть компонентов матрицы. Вследствие этого мешающие влияния посторонних компонентов при электротермической атомизации ниже, чем при пламенной [5]. Анализ твердых образцов. При использовании электротермической атомизации существует принципиальная возможность (при проведении соответствующей градуировки) непосредственного анализа твердых образцов (например, биологических тканей или частиц минералов) [5]. Электротермический способ атомизации требует наличия специальных устройств для очень быстрого нагрева печи, применения защитного инертного газа, а графитовые кюветы должны быть изготовлены из сверхчистого графита. Поэтому электротермический способ атомизации более дорогостоящий, чем пламенный [5]. К числу недостатков электротермического способа следует отнести возможность образования в ходе анализа труднолетучих карбидов металлов. Однако этого явления можно избежать, применяя графитовые печи с платформами. Примером может служить изображенная на рис. 6 графитовая трубка со вставленной в нее пластинкой из тантала.
Рис. 6. Графитовый атомизатор с платформой. Графитовая печь служит в этом случае исключительно для нагрева, а процесс атомизации происходит на платформе. Применяют и печи с платформами, сделанными из графита. Преимущество таких печей по сравнению с обычными состоит в более равномерном нагревании пробы. При этом воспроизводимость результатов анализа значительно улучшается [5]. 1.2.5 Монохроматоры В атомно-абсорбционной спектроскопии роль монохроматора заключается в отсечении лишних линий испускания лампы с полым катодом, молекулярных полос и постороннего внешнего излучения. Диапазон длин волн, представляющий интерес для атомно-абсорбционной спектрометрии, простирается от 193,7 нм (резонансная линия аргона) до 851 нм (линия, используемая для определения цезия). Ввиду слишком широких спектральных полос пропускания использование светофильтров в атомно-абсорбционной спектрометрии невозможно. Его основные детали - это щели, линзы зеркала и диспергирующие элементы, которые разлагают излучение в спектр – дают раздельное изображение спектральных линий (призмы из стекла и кварца и дифракционные решетки). Обычно для монохроматизации используют дифракционные решетки, содержащие 500-3000 штрихов на миллиметр (общее число штрихов достигает при этом порядка 105 ), обратная линейная дисперсия таких решеток составляет от 0,5 до 5 нм/мм. Но так же используются и призмы из стекла в видимом и инфракрасном участке спектра, кварцевые призмы – в УФ области спектра [1].
1.2.6 Детекторы Детектор – приемник света - преобразует падающую на него световую энергию в электрический сигнал. В атомно-абсорбционном анализе для этой цели используют фотоэлектронные умножители (ФЭУ). В них поглощение света либо приводит к отрыву электрона с облучаемой поверхности, либо к увеличению электрической проводимости под действием света. В ходе программируемого нагревания электротермического атомизатора происходит непрерывная регистрации атомно-абсорбционного сигнала во времени [1]. 1.2.7 Количественный анализ Атомно-абсорбционный спектральный анализ — метод количественного анализа, основанный на поглощении электромагнитного излучения атомами анализируемого вещества [6]. В результате неупругого взаимодействия внешнего (диагностирующего) излучения с атомами определенная доля этого излучения поглощается. Таким образом, интенсивность падающего излучения при прохождении через слой вещества уменьшается [6]. В результате поглощения излучения атомы переходят в возбужденные энергетические состояния. Таким переходам в атомных спектрах поглощения соответствуют так называемые резонансные линии. Энергия резонансных переходов и соответствующие частоты поглощения являются строго специфичными для атомов каждого химического элемента. В соответствии с величиной энергии диагностирующего излучения, используемой в атомно-абсорбционной спектрометрии, в атомах анализируемого вещества происходит возбуждение внешних электронов [6]. Для изучения поглощения используются оптические приборы - спектрофотометры. Применение атомно-абсорбционной спектроскопии в количественном химическом анализе основано на существовании взаимно однозначного соответствия между величиной поглощения и концентрацией атомов анализируемого вещества. Это соотношение определяется следующими законами светопоглощения: Закон Бера: каждая частица (атом или молекула) поглощает одну и ту же долю энергии излучения. Тогда коэффициент поглощения k пропорционален количеству поглощающих частиц, которое, в свою очередь, определяет концентрацию анализируемого вещества: k(λ) = α(λ)С (9) Размерность коэффициента α(λ), как следует из (9), обратна размерности концентрации С. Эта величина зависит от длины волны падающего излучения, которая определяет энергию электронного возбуждения и вероятности переходов между электронными уровнями. В свою очередь, совокупность переходов между энергетическими уровнями создает спектр поглощения I(ν) или I(λ), зависящий от длины волны [6]. Закон Бера (9) справедлив для идентичных частиц поглощающей среды. Если, например, при взаимодействии атомов А образуется димеры А2 , происходит или ионизация атомов, или образование молекул при взаимодействии с молекулами или атомами среды, то количество поглощающих частиц уменьшается. Это приводит к кажущемуся отклонению от закона Бера [6]. Закон Бугера—Ламберта: если среда однородна и слой вещества перпендикулярен падающему параллельному (коллимированному) световому потоку, то интенсивность I(λ) уменьшается с увеличением толщины поглощающего слоя по экспоненциальному закону: I = I0 ехр(-kl),(10) где I0 и I — интенсивности падающего и прошедшего излучения; l — толщина слоя вещества. Отклонения от закона Бугера—Ламберта известны только для световых потоков очень большой интенсивности, например, мощного лазерного излучения. Для источников света, используемых в аналитических приборах атомно-абсорбционной спектрометрии, данный закон выполняется с достаточной точностью [6]. Объединяя уравнения (9) и (10), получим формулу для основного закона поглощения света — закона Бугера—Ламберта—Бера: I = I0 ехр(-αCl) (11) В спектральном анализе используется величина относительного пропускания света: T = Эту безразмерную величину часто выражают в процентах. Линейное соотношение между концентрацией и поглощением получаем, если ввести A = -lgT = lg Безразмерную величину A = lg называют абсорбцией поглощающего слоя, где ε = 0,4343α. Если величина концентрации выражена в молях на литр, то ε называют коэффициентом молярного поглощения. Часто используемая размерность ε, моль-1 · л ∙ см-1 . Градуировочный график, построенный в координатах (A,C), представляет прямую, проходящую через начало координат [6]. Чувствительность метода не зависит от концентрации и определяется величиной молярного поглощения ε(λ): S = Таким образом, анализ следует проводить на длине волны λmax излучения, соответствующей максимуму ε(λ). Так же, как и в методе атомно-эмиссионной спектроскопии, в атомно-абсорбционной спектрометрии анализируемое вещество должно находиться в атомизированном состоянии. Однако в отличие от атомно-эмиссионной спектроскопии здесь не требуются высокие температуры, приводящие к электронному возбуждении атомов анализируемого вещества с последующей спонтанной эмиссией излучения. Напротив, в атомно-абсорбционной спектрометрии диагностирующее излучение вызывает резонансное электронное возбуждение свободных атомов, находящихся в основном состоянии. Доля атомов, находящихся в основном состоянии, является преобладающей. При неизменных условиях атомизации исследуемого вещества концентрация атомов в атомизаторе пропорциональна истинной концентрации определяемого элемента в пробе. Тогда для поглощающего слоя можно записать: A = KlC (16) где коэффициент K включает коэффициенты молярного поглощения ε(λ) [см. формулу (14)] и перехода от истинной концентрации определяемого элемента в пробе к его концентрации в атомизаторе, l - толщина светопоглощающего слоя (пламени); C – концентрация [6]. Таким образом, так же как и в атомно-эмиссионной спектроскопии, коэффициент K в формуле (16) находится опытным путем при проведении серии измерений для стандартных образцов исследуемого вещества [6]. В практике анализа обычно применяют метод градуировочного графика и метод добавок. В методе градуировочного графика измеряют оптическую плотность нескольких стандартных растворов и строят график в координатах оптическая плотность - концентрация. Затем в тех же условиях определяют оптическую плотность анализируемого раствора и по градуировочному графику находят его концентрацию [4]. При работе по методу добавок сначала измеряют оптическую плотность анализируемого раствора (Ах ), затем вводят в анализируемый раствор определенный объем стандартного раствора и снова измеряют оптическую плотность (Ах+ст ), Если Cх - концентрация анализируемого раствора, а Cст - стандартного, то Ах = KlCx ; (17) Ах+ст = Kl (Cx + Cст ) (18) Учитывая, что K и l одинаковы, получаем: Ах
Ах+ст - Ах
Метод применим для систем, подчиняющихся в исследуемой области концентраций уравнению: A = lg
1.2.8 Факторы, влияющие на величину абсорбционного сигнала Многие явления могут вызвать искажение величины абсорбционного сигнала. Испускание света возбужденными атомами, флуоресценция атомов или неспецифическое излучение самого атомизатора уменьшают регистрируемую величину светопоглощения. Это приводит к снижению чувствительности определения, повышает предел обнаружения. Для устранения помех, вызванных испусканием света, излучение лампы с полым катодом модулируют с частотой около 100 Гц. При помощи соответствующей настройки приемника излучения (частотной и фазовой фильтрации) на желаемой длине волны можно удалить немодулируемую часть излучения [5]. Рассеяние света и неспецифическое поглощение излучения молекулами (фоновое поглощение) увеличивают регистрируемую величину поглощения. Рассеяние света происходит в первую очередь на твердых, не испарившихся частицах, присутствующих в атомизаторе. В соответствии с законом Релея интенсивность рассеянного излучения пропорциональна третьей степени радиуса частиц и обратно пропорциональна четвертой степени длины волны:
где Is — интенсивность рассеянного излучения, N — число частиц, r — радиус частицы, υ — объем частицы. При переходе от 800 к 200 нм интенсивность рассеянного света при прочих равных условиях возрастает в 256 раз. На практике это означает необходимость особенно тщательной пробоподготовки при работе в коротковолновой области [5]. Молекулярные спектры поглощения отличаются от атомных наличием весьма широких полос — от нескольких нанометров до 100 нм и более. Влияние поглощения света молекулами фона можно устранить различными способами — используя источник непрерывного спектра излучения (дейтериевую лампу), метод “двух линий” или эффект Зеемана [5]. 1.2.8.1 Коррекция фонового поглощения 1.2.8.1.1 Применение дейтериевой лампы В этом методе для коррекции фонового поглощения используют два источника света с различной спектральной шириной светового потока: узкополосные, дающие линейчатый спектр (лампы с полым катодом или безэлектродные разрядные лампы) и широкополосные, дающие непрерывный спектр (дейтериевая лампа – баллон, наполненный D2 , в котором расположены нагреваемый катод, металлический анод и ограничительная апертура между ними; при токе в несколько сот мА газ возбуждается настолько, что испускает интенсивное излучение непрерывного спектра в диапазоне длин волн от 190 нм до 400 нм – именно в этой области расположены резонансные линии большинства исследуемых элементов и наиболее выражены эффекты неселективного поглощения) [7]. Поглощение, измеренное при длине волны резонансной линии ЛПК, складывается из специфического поглощения атомов определяемого элемента и неспецифического поглощения фона. При облучении дейтериевой лампой поглощает только фон. Пробу поочередно облучают световыми потоками ЛПК и дейтериевой лампы (выделяя из нее монохроматором полосу шириной 0,1 - 3 нм) и находят величину специфического (атомного) поглощения А А где I Этот способ коррекции фонового поглощения чрезвычайно распространен. При его использовании следует ясно отдавать себе отчет в его возможностях и границах применимости. Поскольку измерения потоков производятся поочередно, колебания оптических характеристик среды могут привести к погрешностям. Кроме того, использование дополнительного источника света и устройства для переключения световых потоков (прерывателя) увеличивает отношение сигнал-шум. Дейтериевая коррекция фона возможна лишь при длинах волн не выше 400 нм, поскольку при больших длинах волн интенсивность излучения дейтериевой лампы резко падает [5]. 1.2.8.1.2 Метод двух линий Метод двух линий позволяет устранить мешающее влияние фона путем одновременного измерения поглощения при двух длинах волн. В этом методе рядом с резонансной линией поглощения атома выбирают еще одну линию испускания ЛПК, при длине волны которой поглощает только фон. Величину поглощения, измеренного при этой длине волны, используют для коррекции. Выбор такой линии, поглощение при длине волны которой не зависело бы от концентрации определяемого элемента и соответствовало фоновому поглощению при длине волны резонансной линии, может представлять проблему. Кроме того, для одновременного измерения поглощения при двух длинах волн необходим спектрометр специальной конструкции [5]. 1.2.8.1.3 Применение эффекта Зеемана Еще один способ коррекции фонового поглощения в атомно – абсорбционной спектрометрии основан на использовании эффекта Зеемана. Этот способ особенно эффективен в случае сильного фонового поглощения возникающего, например, при анализе биологических образцов. Эффект Зеемана состоит в расщеплении электронных уровней атома под действием магнитного поля [5]. В простейшем случае при действии магнитного поля атомная спектральная линия расщепляется на три близко расположенные линии. Они называются π и σ-компонентами. Положение π-компоненты совпадает с длиной волны исходной линии λ0 , а обе σ-компоненты расположены симметрично относительно нее в области больших и меньших длин волн. При этом излучение π- и σ-компонент по-разному поляризовано: π -компоненты — параллельно, а σ-компонент — перпендикулярно вектору магнитного поля. При пропускании светового потока через поляризационный светофильтр обе компоненты можно разделить. Применяя специальную схему измерения, можно добиться, чтобы поглощение при длине волны π-компоненты соответствовало суммарному специфическому и неспецифическому поглощению, а при длине волны σ-компоненты — только специфическому. Магнитное поле можно налагать на излучатель или атомизатор. Чаще используют второй способ [5].
1.2.9 Помехи и способы их устранения при проведении атомно-абсорбционного анализа 1.2.9.1 Влияния в плямени Одним из основных достоинств атомно-абсорбционного метода является его высокая селективность, тем не менее, метод не свободен от помех, связанных, главным образом, с различием состава градуировочных растворов и реальных проб. Градуировочные растворы в атомной абсорбции обычно представляют собой подкисленные водные растворы определяемых элементов, а в пробе: присутствуют элементы матрицы-катионы и анионы, влияние которых на абсорбцию определяемого элемента может быть значительным [2]. Так же дополнительные ошибки могут закладываться и непосредственно при работе. Так как при работе в пламенном варианте выполняются следующие действия: введение пробы в пламя в виде аэрозоля, испарение анализируемого вещества из частиц аэрозоля, атомизация, взаимодействие свободных атомов с молекулами и радикалами в пламени и ионизация, то и помехи будут связаны с ними [2]. Влияния при получении и переносе аэрозоля. Различные свойства раствора образца могут явиться определяющими для скорости распыления и размера капли: вязкость, поверхностное натяжение, плотность раствора и др. С изменением этих факторов из-за присутствия сопутствующих элементов может существенно измениться скорость подачи анализируемого раствора в пламя, полнота испарения образующихся частиц и, следовательно, величина абсорбции [2]. Однако степень влияния не всегда находится в соответствии с изменением скорости распыления и вязкости. Эти факторы не являются основной причиной изменения аналитического сигнала при увеличении концентрации раствора до 5-10%. В большей степени это влияние связано с природой мешающих соединений (матрицы). Изменение чувствительности определения элемента обусловлено эффектом связанным с образованием соединений, частиц или кристаллов солей большего размера и, соответственно, к затруднению испарения элемента примеси из частиц аэрозоля из-за меньшей летучести крупных кристаллов и частиц [2]. Влияния в конденсированной фазе. Время пробега частицей аэрозоля расстояния от основания горелки до области оптической оси очень мало (порядка 10° с). В присутствии мешающих компонентов, которые химически связывают или физически блокируют испарение определяемого элемента, этого времени оказывается недостаточно для полного протекания процессов испарения и атомизации [2]. В восстановительном воздушно-ацетиленовом пламени и пламени ацетилен-динитроксид возможны влияния, при которых труднодиссоциируемые элементы увеличивают абсорбцию других элементов. Предложено следующее объяснение этого влияния. Элементы, более труднолетучие в металлическом состоянии или в форме карбидов, чем в виде оксидов, определяются в восстановительном пламени с низкой чувствительностью. Это связанно с тем, что еще в твердых аэрозольных частицах происходит восстановление элемента до нелетучих продуктов. Присутствие оксидов других элементов способствует процессу окисления определяемого элемента до более летучего оксида, и испарение аэрозольных частиц преобладает над процессом восстановления [2]. Устранить подобные помехи можно следующим образом: 1. Оптимизация аппаратурных параметров. а) Высота светового пучка над горелкой определяет время пробега частицы, поэтому при большей рабочей высоте пламени влияния уменьшаются. б) При определении многих элементов в окислительном пламени влияния уменьшаются или полностью устраняются благодаря высокой температуре пламени и отсутствию преждевременного восстановления и карбидообразования в конденсированной фазе. 2. Повышение дисперсности аэрозоля. 3. Использование высокотемпературных пламен. 4. Введение специальных добавок - спектрохимических буферов. Буфер вводят как в раствор проб, так и в градуировочные растворы [2]. Влияния в газовой фазе. В высокотемпературном пламени наиболее часто играет роль помеха, связанная с ионизацией определяемого элемента. Для достижения максимальной чувствительности и правильности анализа необходимо, чтобы степень Ионизации была минимальной и одинаковой для градуировочных растворов и проб [2]. Степень ионизации зависит от: · температуры пламени; · потенциала ионизации определяемого элемента; · концентрации определяемого элемента в анализируемом растворе; · содержания посторонних компонентов и, прежде всего, легкоионизи-руемых элементов в пробе. На степень ионизации определяемого элемента оказывает также влияние стехиометрия и рабочая высота пламени. Ионизационные помехи проявляются: 1. в пламени пропан-воздух: только при определении рубидия и цезия; 2. в пламени воздух-ацетилен; при определении всех щелочных металлов, бария и кремния; 3. в пламени динитроксид-ацетилен: при определении почти всех элементов: полностью ионизируются щелочные, в значительной степени- щелочноземельные, частично-редкоземельные элементы и алюминий, галлий, индий и теллур и незначительно - медь, кремний, титан, свинец, хром, молибден, вольфрам и др. [2]. Ионизацию можно подавить добовлением в анализируемый и градуиро-вочные растворы ионизационного буфера. Для этой цели используют хлориды калия, цезия, лития и лантана в концентрации 0.1-1 % [2]. Спектральные помехи. Наложение и перекрытие резонансных линий различных элементов в атомно-абсорбционной спектрометрии встречается очень редко. Тем не менее, необходимо отметить для примера ряд таких случаев (первым в паре указан определяемый элемент, вторым - мешающий): · линия 242.79 нм золота и линия 242.82 нм железа; · линия 253.65 нм ртути и - 253.65 нм кобальта (при определении ртути · методом холодного пара кобальт не мешает); · линия 324.75 нм меди и - 324.75 нм европия. При измерении концентрации одного из элементов такой пары другой будет играть роль спектральной помехи, если он находится в большом избытке. Устранить такую помеху можно либо отделением мешающего компонента, либо использованием другой спектральной линии определяемого элемента [2]. 1.2.9.2 Методы устранения мешающих влияний в пламенном атомно-абсорбционном анализе 1. Разбавление раствора – ослабляет действие всех мешающих влияний, кроме ионизационных, однако неэффективно при сильных химических влияниях. 2. Уравнивание концентрации основного компонента в градуировочных растворах и пробах. Применяют для учета влияния матричного эффекта в том случае, когда известен состав матрицы проб. Метод используется при работе с органическими растворителями, при введении спектрохимических буферов. Следует поддерживать примерно одинаковым содержанием кислот в растворах пробы и градуировочных. 3. Оптимизация аппаратурных условий. В первую очередь это относится к стехиометрии пламени, рабочей высоте наблюдения. Важными параметрами являются ток лампы, спектральная ширина щели монохроматора. 4. Введение буферов. Наиболее эффективный способ устранения мешающих влияний в конденсированной фазе – это введение «освобождающих» спектрохимических буферов, «испаряющих» и «защитных» добавок или их смесей. 5. Использование высокотемпературного пламени динитроксид-ацетилен. 6. Использование метода добавок. 7. Добавление органических растворителей. 8. Буферирование – введение ионизационного буфера, например, КС1; введение в пробы и градуировочные растворы избытка мешающегокомпо-нента до насыщения мешающего эффекта. 9. Химическое разделение [2]. 1.2.9.3 Влияния в графитовой печи Рассмотрим источники ошибок при выполнении анализа. 1. Введение пробы. Здесь существенна точность дозировки, на которую влияют изменение вязкости и поверхности натяжения раствора, загрязнение кончика дозатора. Автоматическое дозирование снимает ряд проблем, но требуется точная юстировка дозирующего устройства [2]. 2. Высушивание. На этой стадии наибольшую опасность представляет разбрызгивание раствора, что ведет к плохой воспроизводимости. 3. Разложение. Здесь зависящими от матрицы важными параметрами являются предельно допустимая температура и скорость ее повышения. При этом возможны потери летучих соединений (например, галогенидов в следующих элементов: Li, Сu, Аg, Zn, Сd, А1, Со,Ni и др.) [2]. 4. Атомизация. Механизм атомизации зависит от химических и физических свойств определяемого элемента и его соединений, от наличия примесей, температурной программы и др. Атомизации предшествуют различные процессы: сублимация, испарение летучих соединений, диссоциирующих в газовой фазе, восстановление оксидов на поверхности графита с последующим испарением металла и т. д. Различия в летучести и термической устойчивости соединений определяемого элемента в пробах и градуировочных растворах могут привести к существенным помехам. Так как температура газовой фазы ниже температуры графита печи и время пребывания вещества в этой фазе невелико, то необходимо, чтобы проба находилась в контакте с поверхностью графитовой печи как можно дольше. Поэтому наличие галогенидов в исходной пробе занижает результат. Снижение аналитического сигнала может быть связано и с труднолетучей матрицей (фосфаты, сульфаты и др.) [2]. 5. Высокотемпературная очистка служит для устранения остатков нелетучих соединений и определяемого элемента, чтобы избежать загрязнения вводимых при последующих измерениях проб [2]. 1.2.9.4 Способы устранения мешающих влияний 1. Оптимизация температурной программы для каждой матрицы; использование максимальной скорости разогрева трубки. 2. Использование платформы. 3. Модификация матрицы. Компоненты матрицы можно превратить в удобную для атомизации форму введением добавок-модификаторов. Например, легколетучие элементы (Аs, Вi, Sе, Те) переводят в труднолетучие соединения добавлением солей никеля или палладия. Допустимая температура разложения может быть повышена при этом до 1000° - 1200°С. При этой температуре могут быть устранены мешающие компоненты и уменьшено неселективное поглощение. Модификация матрицы может использоваться и для удаления некоторых компонентов путем перевода их в легколетучие соединения, например, для удаления NaС1 добавляют NH4 NO3 . Модификатор может играть роль диспергатора, пространственно разделяя определяемый и мешающий компоненты. В роли диспергатора могут выступать, например, аскорбиновая и щавелевая кислоты [2]. 4. Использование Зеемановской коррекции фона. 5. Регистрация интегральной абсорбции устраняет помехи, связанные с различной скоростью испарения определяемого элемента из матрицы и гра-дуировочных растворов, улучшает линейность градуировочного графика. 6. Использование метода добавок [2]. 1.2.10 Метрологические характеристики метода Чувствительность . Пределы обнаружения в атомно-абсорбционной спектрометрии для большинства элементов составляют 10-6 – 10-4 в пламенном и 10-9 – 10-7 % масс, в электротермическом вариантах. Таким образом, атомно-абсорбционная спектрометрия в целом чувствительнее, чем атомно-эмиссионная спектроскопия. Это связано с тем, что в атомно-абсорбционной спектрометрии аналитический сигнал формируют атомы, находящиеся в основном состоянии, т. е. основная доля атомов определяемого элемента, а в атомно-эмиссионной спектроскопии — атомы в возбужденном состоянии, доля которых весьма мала [8]. Диапазон определяемых содержаний в атомно-абсорбционной спектрометрии лимитируется величиной аналитического сигнала (оптической плотности А), который можно измерить с необходимой точностью. Диапазон значений обычно составляет от нескольких сотых до 0,6 – 1,2 единиц оптической плотности. Таким образом, диапазон содержаний, определяемых методом атомно-абсорбционной спектрометрии, не превышает 1 – 2 порядка величин. Проблемы с определением малых значений А связаны со способом измерения оптической плотности — по разности между интенсивностями падающего и прошедшего излучений. При малых оптических плотностях эта разность мала и погрешность, соответственно, велика. В областях высоких оптических плотностей погрешности связаны, главным образом, с существенными отклонениями от основного закона светопоглощения, вызванными недостаточной монохроматичностью излучения источника и влиянием рассеянного света, а также неоднородностью поглощающей среды (атомного пара, имеющего различную концентрацию в различных областях пространства атомизатора). Малый диапазон определяемых содержаний является существенным недостатком метода атомно-абсорбционной спектрометрии [1]. Воспроизводимость в атомно-абсорбционной спектрометрии (особенно в пламенном варианте), как правило, несколько выше, чем в атомно-эмиссионной спектроскопии. Величины sr обычно составляют 0,005 – 0,05 для пламенного и 0,02 – 0,10 для электротермического способов атомизации. Улучшение воспроизводимости для атомно-абсорбционной спектрометрии по сравнению с атомно-эмиссионной спектроскопией связано в первую очередь с тем, что флуктуации температуры атомизатора почти не изменяют долю невозбужденных атомов (она всегда близка к 100%), однако сильно влияют на долю возбужденных атомов (в соответствии с уравнением Больцмана) [8]. Систематические ошибки атомно-абсорбционного анализа связаны, в основном, с несоответствием стандартных образцов и проб, которое обусловлено: 1) наложением линий третьего элемента на линию определяемого (0,05%); 2) рассеивание света и населективностью поглощения, которое устраняется повышением температуры атомизации (0,02%); 3) химическим воздействием на определяемый элемент каким-либо другим элементом, и как следствие, уширение линии поглощения определяемого элемента (0,03%) [1]. Случайные ошибки атомно-определяемого определения связаны с нестабильностью источников света, атомизатора, детектора, а также связаны с точностью измерительной шкалы, снизить эти ошибки можно, работая в оптимальном интервале абсорбционности [1]. Селективность в атомно-абсорбционной спектрометрии часто бывает выше, чем в атомно-эмиссионной спектроскопии. Это объясняется тем, что в атомно-абсорбционной спектрометрии практически никакой роли не играет наложение спектральных линий. Селективность в атомно-абсорбционной спектрометрии лимитируется, главным образом, не спектральными, а физико-химическими помехами, которые возможно устранить [8]. Главный недостаток метода атомно-абсорбционной спектрометрии — трудность осуществления многоэлементного анализа, поскольку для каждого элемента нужен свой источник излучения. По этой же причине метод атомно-абсорбционной спектрометрии непригоден для качественного анализа [8]. Количественный анализ. Метод атомно-абсорбционной спектрометрии — один из наиболее чувствительных и удобных методов массовых одноэлементных определений большинства металлов. Для количественного анализа методом атомно-абсорбционной спектрометрии применяют методы внешних стандартов (градуировочного графика) и добавок. Метод внутреннего стандарта, в отличие от атомно-эмиссионной спектроскопии, неприменим ввиду того, что атомно-абсорбционная спектрометрия — одноэлементный метод анализа, не позволяющий одновременно измерять аналитические сигналы двух элементов — определяемого и внутреннего стандартов. Особенно широко в атомно-абсорбционной спектрометрии используют метод добавок. Это связано с тем, что помехи в атомно-абсорбционной спектрометрии имеют главным образом физико-химическую природу, т. е. являются с метрологической точки зрения мультипликативными. Кроме того, атомно-абсорбционной спектрометрия — это главным образом метод анализа растворов. Для растворов, в отличие от твердых проб, метод добавок легко реализуем технически [8]. Наиболее оптимальные спектральные параметры атомно-абсорбционного определения некоторых элементов представлены в таблице № 4. Таблица №4. Спектральные параметры атомно-абсорбционного определения некоторых элементов.
1.2.11 Область практического применения ААС Атомно-абсорбционные спектрометры применяются в экологии, геологоразведке, контроле технологических процессов, производственной санитарии, научных исследованиях. Экологический контроль: измерение содержания различных элементов в воде, почве, донных отложениях, атмосферном воздухе, а также тканях растительного и животного происхождения. Технологический контроль: · экспресс-анализ и непрерывный контроль состава веществ в технологических процессах; · входной контроль, контроль готовой продукции. Медицина: анализ тканей и жидкостей биологического происхождения (кровь, моча, волосы и др.). Криминалистика: идентификация примесей и следовых количеств элементов. Ветеринарные лаборатории: корма, кровь, продукты животноводства. Контролирующие и сертифицирующие лаборатории: анализ пищевых продуктов и кормов, анализ сточных, природных, питьевых вод и воздуха. Контроль качества вин и крепких напитков [9]. 1.3 Определение примесей в марганце различными методами анализа
1.3.1 Фотометрическое определение фосфора в металлическом и азотированном марганце Гост 16698.4-93 устанавливает фотометрические методы определения фосфора при массовой доле его в металлическом и азотированном марганце от 0,002 до 0,09 %. 1.3.1.1 Фотометрический метод с применением аскорбиновой кислоты. Сущность метода. Метод основан на образовании фосфорно-молибденовой гетероноликислоты с последующим восстановлением ее аскорбиновой кислотой в присутствии сурьмяно-винно-кислого калия до комплексного соединения, окрашенного в синий цвет и измерении его оптической плотности. Область применения. Определение фосфора при массовой доле его в металлическом и азотированном марганце от 0,002 до 0,09 %. Обработка результатов. Массовую долю фосфора Х, % вычисляют по формуле:
где m1 – масса фосфора, найденная по градуировочному графику, г; m – масса навески, соответствующая аликвотной части раствора пробы, г. Точность.
1.3.1.2 Фотометрический метод с применением тиомочевины или ионов двухвалентного железа Сущность метода. Метод основан на реакции образования желтой фосфорно-молибденовой гатерополикислоты с последующим восстановлением в соляно-кислой среде тиомочевинной или ионами двухвалентного железа в присутствии соляно-кислого гидроксиламина до соединения, окрашенного в синий цвет, и измерении его оптической плотности. Область применения. Метод применяется при массовой доле фосфора от 0,01 до 0,9%. Обработка результатов. Метод применяется при массовой доле фосфора от 0,01 до 0,9%. 1)Массовую долю фосфора Х1 , %, определяемую методом градуировочного графика, вычисляют по формуле:
где m– масса фосфора, найденная по градуировочному графику, г; m1 – масса навески, соответствующая аликвотной части раствора пробы, г. 2) Массовую долю фосфора Х2 , %, определяемую методом сравнения, вычисляют по формуле:
где А – аттестованное значение массовой доле фосфора в стандартном образце, %; D – значение оптической плотности раствора пробы; D1 – значение оптической плотности раствора контрольного раствора контрольного опыта; D2 – значение оптической плотности раствора стандартного образца. 1.3.2 Фотометрическое и гравиометрическое определение кремния в металлическом и азотированном марганце ГОСТ 16698.5-71 устанавливает методы определения кремния в металлическом и азотированном марганце: фотометрический при массовой доле его от 0,1 до 2,5% и гравиметрический при массовой доле его от 0,5 до 2,5%. 1.3.2.1 Фотометрическое определение кремния в металлическом и азотированном марганце Сущность метода. Метод основан на преобразовании желтой кремнемолибденовой гетерополикислоты с последующим восстановлением ее аскорбиновой кислотой до комплексного соединения, окрашенного в синий цвет, и измерении его оптической плотности. Область применения. Метод применяется при массовой доле кремния в пробе от 0,1 до 2,5 %. Обработка результатов. Массовую долю кремния X, %, определяемую по градуировочному графику и вычисляют по формуле:
где m1 – масса кремния, найденного по градуировочному графику, г; m - масса навески, соответствующая аликвотной части раствора пробы, г. Массовую долю кремния Х1 , %, определяемую методом сравнения, вычисляют по формуле:
где А – аттестованное значение массовой доли кремния в стандартном образце, %; D - оптическая плотность раствора пробы; D1 - оптическая плотность раствора контрольного опыта; D2 - оптическая плотность раствора стандартного образца. Точность.
1.3.2.1 Гравиметрический метод кремния в металлическом и азотированном марганце Сущность метода. Метод основан на выделении из хлорно-кислого раствора кремния в виде кремниевой кислоты, прокаливании ее до диоксида кремния и удалении в виде тетрафорида кремния. Область применения. Метод применяется при массовой доле кремния в пробе от 0,5 до 2,5 %. Обработка результатов. Массовую долю кремния X2 , %, определяемую по формуле:
где m1 – масса тигля с осадком диоксида кремния до обработки фтористоводородной кислотой, г; m2 – масса тигля с осадком диоксида кремния после обработки фтористоводородной кислотой, г; m3 - масса тигля с остатком контрольного опыта до обработки фтористоводородной кислотой, г; m4 - масса тигля с остатком контрольного опыта после обработки фтористоводородной кислотой, г; m - масса навески пробы, г; 0,4674 – коэффициент пересчета диоксида кремния на кремний. 1.3.3 Фотометрическое, атомно-абсорбционное и титриметрическое определения железа в металлическом и азотированном марганце ГОСТ 16698.6-71 устанавливает методы определения железа в металлическом и азотированном марганце: фотометрические и атомно-абсорбционный при массовой доле железа от 0,1 до 3,5%; титриметрический при массовой доле железа от 0,5 до 3,5%. 1.3.3.1 Фотометрическое определение железа в металлическом и азотированном марганце с применением 1,10-фенантроина Сущность метода. Метод основан на образовании комплексного соединения двухвалентного железа с 1,10-фенантролином, измерении его оптической плотности на спектрофотометре при длине волны 510 нм или на фотоэлектроколориметре в диапазоне длин волн от 480 до 530 нм. Область применения. Метод применяется при массовой доле железа от 0,1 до 3,5%. Обработка результатов. Массовую долю железа Х, %, вычисляют по формуле:
где m1 – масса железа, найденная по градуировочному графику, г; m - масса навески, соответствующая аликвотной части раствора пробы, г. Точность.
1.3.3.2 Фотометрический метод с применением сульфосалициловой кислоты Сущность метода. Метод основан на образовании желтого комплексного соединения железа с сульфосалициловой кислотой в щелочной среде (рН 8-12) и измерении его оптической плотности на спектрофотометре при длине волны 430 нм. Область применения. Метод применяется при массовой доле железа от 0,1 до 3,5%; Обработка результатов. Массовую долю железа Х, %, вычисляют по формуле:
где m1 – масса железа, найденная по градуировочному графику, г; m - масса навески, соответствующая аликвотной части раствора пробы, г. Точность. Описана в фотометрическом методе. 1.3.3.3 Атомно-абсорбционный метод Сущность метода. Метод основан на измерении атомной абсорбции железа в пламени воздух-ацителен при длине волны 248,4 нм. Навеску пробы предварительно растворяют в смеси кислот. Область применения. Метод применяется при массовой доле железа от 0,5 до 3,5%. Обработка результатов. Массовую долю железа Х, %, определяемую по градуировочному графику и вычисляют по формуле:
где m1 – масса железа, найденная по градуировочному графику, г; m - масса навески, соответствующая аликвотной части раствора пробы, г. Массовую долю железа Х1 , %, определяемую методом сравнения, вычисляют по формуле:
где А – аттестованное значение массовой доли кремния в стандартном образце, %; D - оптическая плотность раствора пробы; D1 - оптическая плотность раствора контрольного опыта; D2 - оптическая плотность раствора стандартного образца. Точность. Описана в фотометрическом методе. 1.3.3.4 Титраметрический метод Сущность метода. Метод основан на восстановлении трехвалентного железа раствором двухлористого олова с последующим титрованием двухвалентного железа двухромовокислым калием с индикатором дифиниламинминосульфонатом натрия. Область применения. Метод используется при массовой доле железа от 0,5 до 2,5%. Обработка результатов. Массовую долю железа Х, %, определяемую по формуле:
где с – массовая концентрация раствора двухромово-кислого калия, г/см3 ; V1 – объем раствора двухромово-кислого калия, израсходованного на титрования раствора пробы, см3 ; V2 - объем раствора двухромово-кислого калия, израсходованного на титрования контрольного опыта, см3 ; m – масса навески пробы, г. Точность. Описана в фотометрическом методе. 1.3.4 Фотометрическое и атомно-абсорбционное методы определения никеля в металлическом марганце и металлическом азотированном марганце ГОСТ 16698.7-71 устанавливает фотометрический и атомно-абсорбционный методы определения никеля при массовой доле никеля в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%.
1.3.4.1 Фотометрическое определениея никеля в металлическом марганце и металлическом азотированном марганце. Сущность метода. Метод основан на образовании окрашенного в вино-красный цвет растворимого комплексного соединения никеля с диметилглиоксимом в щелочной среде и присутствии окислителя – сернокислого аммония и измерении оптической плотности раствора на спектрофотометре при длине волны 530 нм или фотоэлектрофотометре в диапазоне длин волн от 530 до 550 нм. Предварительно проводят определение никеля от больших количеств марганца путем фракционного осаждения его совместно с железом в виде сульфидов. Область применения. Определение проводится при массовой доле никеля в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%. Обработка результатов:
где m1 – масса никеля, найденная по градуировочному графику, г; m – масса навески, соответствующая аликвотной части раствора, г. 1.3.4.2 Атомно-абсорбционное определение никеля в металлическом марганце и металлическом азотированном марганце Сущность метода. Метод основан на растворении навески в хлорной и фтористоводородной кислотах, распылении раствора в пламя воздух-ацитилен и измерении атомной абсорбции никеля при длине волны 232 нм. Область применения. Определение проводится при массовой доле никеля в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%. Обработка результатов:
где С –массовая концентрация никеля в растворе анализируемой пробы, найденная по градуировочному графику г/см3 ; v – общий объем раствора, см3 ; m – масса навески, г. 1.3.5 Фотометрическое и атомно-абсорбционное методы определения меди в металлическом марганце и металлическом азотированном марганце ГОСТ 16698.9-71 устанавливает фотометрический и атомно-абсорбционный методы определения меди при массовой доле меди в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%. 1.3.5.1 Фотометрическое определение меди в металлическом марганце и металлическом азотированном марганце. Сущность метода. Метод основан на образовании в аммиачной среде при pH=9 окрашенного в бурый цвет комплексного соединения меди с диэтилдитиокарбамидоматом натрия. От всех мешающих элементов медь предварительно отделяют в слабокислом растворе осаждением серноватистокислым натрием. По интенсивности окраски комплекса находят содержание меди. Область применения. Определение проводится при массовой доле меди в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%. Обработка результатов:
где g – количество меди, найденное по калибровочному графику, мг.; G – навеска, соответствующая аликвотной части раствора, г.. Точность.
1.3.5.2 Атомно-абсорбционное определение меди в металлическом марганце и металлическом азотированном марганце Сущность метода. Метод основан на растворении навески в хлорной и фтористоводородной кислотах, распылении раствора в пламя воздух-ацитилен и измерении атомной абсорбции никеля при длине волны 327,7 нм. Область применения. Определение проводится при массовой доле меди в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,05%. Обработка результатов:
где С –массовая концентрация меди в растворе анализируемой пробы, найденная по градуировочному графику, г/см3 ; v – общий объем раствора, см3 ; m – масса навески, г. Точность.
1.3.6 Атомно-абсорбционное методы определения кальция и магния металлическом марганце и металлическом азотированном марганце ГОСТ 16698.12-84 устанавливает атомно-абсорбционный метод определения кальция и магния при их массовой доле в металлическом марганце и металлическом азотированном марганце от 0,10 до 0,70% каждого. Сущность метода. Метод основан на растворении навески в хлорной и фтористоводородной кислотах, распылении раствора в пламя воздух-ацитилен или закись азота-ацителена и измерении атомной абсорбции кальция при длине волны 422,7 нм, магния при длине волны 285,2 нм. Область применения. Метод применяется при их массовой доле в металлическом марганце и металлическом азотированном марганце от 0,10 до 0,70% каждого. Обработка результатов:
где С –массовая концентрация кальция (магния) в растворе анализируемой пробы, найденная по градуировочному графику г\см3 ; v – общий объем раствора, см3 ; m – масса навески, г. Точность.
1.3.7 Фотометрический метод определения алюминия в металлическом марганце и металлическом азотированном марганце ГОСТ 16698.10-71 устанавливает фотометрический метод определения алюминия при массовой доле алюминия в металлическом марганце и металлическом азотированном марганце от 0,02 до 0,07 %. Сущность метода. Метод основан на образовании в ацетатнобуферной среде окрашенного в красный цвет внутрикомплексного соединения алюминия с алюминоном. По интенсивности окраски комплекса находят содержание алюминия. Мешающие определению элементы предварительно отделяют осаждением избытком щелочи. Область применения. Метод применяется при массовой доле алюминия в металлическом марганце и металлическом азотированном марганце от 0,02 до 0,07 %. Обработка результатов. Содержание алюминия в пробе (Х) в процентах вычисляют по формуле:
где д – количество алюминия, найденного по калибровочному графику, в мг.; G – навеска, соответствующая аликвотной части раствора, в г. Точность.
1.3.8 Фотометрический метод определения титана в металлическом марганце и металлическом азотированном марганце ГОСТ 16698.11-71 устанавливает фотометрический метод определения титана при массовой доле титана в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,10%. Сущность метода. Метод основан на образовании окрашенного в желто-оранжевый цвет комплексного соединения титана с диантипирилмэтаном. По интенсивности окраски комплекса находят содержание титана. Титан предварительно выделяют купферроном. Мешающие влияние трехвалентного железа и пятивалентного ванадия устраняют прибавлением аскорбиновой кислоты. Область применения. Метод применяется при массовой доле титана в металлическом марганце и металлическом азотированном марганце от 0,005 до 0,10 %. Обработка результатов. Содержание титана в пробе (Х) в процентах вычисляют по формуле:
где д – количество титана, найденного по калибровочному графику, в мг.; G – навеска, соответствующая аликвотной части раствора, в г. Точность.
1.3.9 Определение марганца в металлическом и азотированном марганце ГОСТ 16698.1-93 устанавливает потенциометрический метод определения марганца в металлическом и азотированном марганце при массовой доле марганца от 80,0 до 96,5 %. При массовой доле выше 96,5 % определение марганца проводится по разности. Сущность метода. Метод основан на окислении двухвалентного марганца до трехвалентного раствором марганцево-кислого калия в нейтральной среде в присутствии комплексообразователя – пирофосфорного кислого натрия или калия. Область применения. Метод применяется при массовой доле марганца в металлическом и азотированном марганце от 80,0 до 96,5 %. Обработка результатов. Массовую долю марганца Х, %, вычисляют по формуле:
где с – массовая концентрация раствора марганцево-кислого калия или натрия г\см3 ; V2 – объем раствора марганцево-кислого калия или натрия, израсходованного на титрования раствора пробы, см3 ; V3 - объем раствора марганцево-кислого калия или натрия, израсходованного на титрования контрольного опыта, см3 ; m – масса навески пробы, г. Точность.
2. ЭКСПЕРЕМЕНТАЛЬНАЯ ЧАСТЬ
2.1 Приборы, оборудование и реактивы
1. Атомно-абсорбционный спектрометр GBC “AVANTA PM” (Австралия). 2. Марганец марки Мн 998. 3. Кислота азотная по ГОСТ 11125-84 квалификации “ос.ч.” и её растворы 0,1М, 1М и 30%. 4. Кислота серная по ГОСТ 14262-78 квалификации “х.ч.” и её растворы 0,1М, 0,5М. 5. Кислота хлороводородная по ГОСТ 14261-77 квалификации “ос.ч.” и её 1М раствор. 6. Государственный стандартный образец состава водного раствора ионов алюминия ГСО 8059-2004. Материал ГСО представляет собой водный раствор ионов алюминия, подкисленный 0,1 М серной кислотой. Аттестованное значение массовой концентрации ионов алюминия 1,000 мг/см3 . 7. Государственный стандартный образец состава водного раствора ионов железа (III) ГСО 7766-2006. Материал ГСО представляет собой раствор железоаммонийных квасцов в 1 М хлороводородной кислоте. Аттестованное значение массовой концентрации ионов железа (III) 1,000 мг/см3 . 8. Государственный стандартный образец состава водного раствора ионов меди ГСО 7764-2006. Материал ГСО представляет собой раствор меди сернокислой в 0,5 М серной кислоте. Аттестованное значение массовой концентрации ионов меди 1,000 мг/см3 . 9. Государственный стандартный образец состава водного раствора ионов никеля ГСО 7785-2006. Материал ГСО представляет собой раствор никеля азотнокислого в 1 М азотной кислоте. Аттестованное значение массовой концентрации ионов никеля 1,000 мг/см3 . 10. Бидистиллированная вода по ГОСТ 6709-72. 11. Колбы мерные 1-го класса точности с притёртой пробкой по ГОСТ 1770-74. 12. Пипетки 1-го класса точности по ГОСТ 29228-91, 29169-91. 13. Стаканы химические по ГОСТ 25336-82. 14. Термометр ртутный по ГОСТ 28498-90. 15. Скарификатор (входит в комплект поставки стандартных образцов). 16. Бумага фильтровальная. 17. Весы аналитические 2-го класса точности. 18. Плита электрическая с закрытой спиралью.
2.2 Подготовка к проведению анализов и приготовление серии стандартных растворов
Для приготовления серии стандартных растворов использовали государственные стандартные образцы состава водного раствора ионов металлов. Подготовка стандартного образца к применению заключалась в приготовлении из него методом объемного разбавления растворов с необходимой массовой концентрацией ионов металла. Приготовления растворов проводили по следующей методике: 1. Обмыли снаружи ампулу со стандартным образцом бидистиллированной водой и высушили поверхность ампулы фильтровальной бумагой. 2. Вскрыли ампулу со стандартным образцом с помощью скарификатора и перелили содержимое в чистый сухой химический стакан. 3. Отобрали из химического стакана чистой сухой пипеткой необходимый объём стандартного образца, VСО , см3 , предварительно рассчитав его по формуле: VСО = 0,001С ∙ Vк / Cm , где C - массовая концентрация ионов металла в приготавливаемом растворе, мг/дм3 ; Cm – аттестованное значение массовой концентрации ионов металла в стандартном образце, г/дм3 . Vк – объем используемой мерной колбы, см3 . Перенесли необходимый объём стандартного образца в мерную колбу, довели объем раствора до метки раствором азотной, либо серной, либо хлороводородной кислоты соответствующей концентрации (в зависимости от состава материала ГСО), колбу закрыли пробкой и тщательно перемешали содержимое колбы [14-20]. Данным методом были приготовлены следующие растворы: Растворы ионов алюминия с массовыми концентрациями 0,300 мг/дм3 , 0,500 мг/дм3 , 0,750 мг/дм3 , 1,000 мг/дм3 , 1,250 мг/дм3 , 1,500 мг/дм3 , 1,750 мг/дм3 , 2,000 мг/дм3 , 2,250 мг/дм3 , 2,500 мг/дм3 . Растворы ионов железа с массовыми концентрациями 0,250 мг/дм3 , 0,500 мг/дм3 , 0,750 мг/дм3 , 1,000 мг/дм3 , 1,250 мг/дм3 , 1,500 мг/дм3 , 1,750 мг/дм3 , 2,000 мг/дм3 , 2,250 мг/дм3 , 2,500 мг/дм3 . Растворы ионов меди с массовыми концентрациями 0,050 мг/дм3 , 0,100 мг/дм3 , 0,150 мг/дм3 , 0,200 мг/дм3 , 0,250 мг/дм3 , 0,300 мг/дм3 , 0,350 мг/дм3 , 0,400 мг/дм3 , 0,450 мг/дм3 , 0,500 мг/дм3 . Растворы ионов никеля с массовыми концентрациями 0,030 мг/дм3 , 0,050 мг/дм3 , 0,100 мг/дм3 , 0,150 мг/дм3 , 0,250 мг/дм3 , 0,300 мг/дм3 , 0,350 мг/дм3 , 0,400 мг/дм3 , 0,450 мг/дм3 , 0,500 мг/дм3 . Растворы из стандартных образцов готовили и использовали при температуре окружающей среды 20˚С. 2.3 Отбор и хранение проб
Для отбора проб использовали сосуды из полиэтилена и боросиликатного стекла, предварительно промытые азотной кислотой, а затем ополоснутые водой. Пипетки и мерные колбы имели 1 класс точности, допустимая погрешность которого представлена в таблице №5. Таблица №5. Допустимая погрешность мерной лабораторной посуды 1-го класса точности [10].
2.4 Приготовление исходных растворов марганца
Навеску металлического марганца 0,5000 г. поместили в термостойкую стеклянную коническую колбу вместимостью 200 см3 , растворили в 50 см3 раствора 30% азотной кислоты (1:1по объему) при нагревании, затем раствор прокипятили до удаления оксида азота. Охлажденный раствор перенесли в мерную колбу вместимостью 500 см3 , долили бидистиллированной водой до метки и перемешали. 2.5 Определение систематической ошибки приготовления растворов марганца
Концентрацию раствора рассчитывали по формуле:
Далее находили относительную погрешность каждой из величин, входящих в вышеприведенную формулу. Относительная погрешность концентрации равна:
Абсолютная погрешность концентрации равна [11]: ∆c = Результаты представлены в таблице №6. Таблица №6. Расчет относительной и абсолютной погрешности концентрации раствора марганца.
Величина систематической ошибки приготовления раствора марганца очень мала, поэтому ей можно пренебречь при расчете погрешностей результатов анализа металлических примесей в марганце. 2.6 Методика определения металлических примесей в образцах марганца марки Мн 998 методом атомно-абсорбционной спектрометрии согласно ГОСТ 16698.6-71, ГОСТ 16698.7-71, ГОСТ 16698.9-71, ГОСТ 16698.10-71 Анализируемые растворы и серию градуировочных растворов (в порядке увеличения концентрации) распыляли в пламя горелки и измеряли атомное поглощение (абсорбцию) определяемых элементов по аналитическим линиям с длинами волн, приведенными в таблице №7. После каждого измерения распылительную систему промывали бидистиллированной водой [6]. Для холостого определения использовали бидистиллированную воду. В данной работе использовали серии градуировочных растворов, приготовленных из водных растворов ГСО. Условия измерения, представленные в таблице №7, подбирали в соответствии с анализируемым элементом. Таблица №7. Условия проведения анализа [10].
Данная модель атомно-абсорбционного спектрометра позволяет проводить измерения в автоматическом режиме. Количественный анализ примесей проводили по методу градуировочного графика. Для построения градуировочной зависимости распыляли холостой раствор и серию градуировочных растворов (в порядке увеличения концентрации). Полученные результаты представлены в таблицах № 8, 11, 14, 17. Представленные в выше указанных таблицах результаты были использованы для построения графиков зависимости значения величины абсорбции от концентрации (мг/дм3 ) металла в стандартных растворах. Расчет параметров линейного уравнения градуировочной зависимости и их доверительных интервалов проводили по формулам, представленным в работе [8]. Градуировочная зависимость абсорбции от концентрации описывается линейным уравнением: Y = ax + b. Используя метод наименьших квадратов коэффициенты уравнения линейной регрессии вычисляли по следующим формулам:
Также были рассчитаны доверительные интервалы для параметров a и b градуировочной зависимости, с применением метода наименьших квадратов. Дисперсия, характеризующая рассеяние экспериментальных значений y для n образцов сравнения относительно рассчитанной прямой Y = ax + b, определяется выражением:
Дисперсии параметров a и b равны:
где Из дисперсий были рассчитаны стандартные отклонения и доверительные интервалы для a и b: sa
= sb
=
где tp - коэффициент Стьюдента. P принимали равным 0,95; Число степеней свободы f = n – 1. При P = 95%, f = 10 – 1 = 9, tp = 2,26 [8].
2.7 Формулы для статистической обработки полученных результатов
По градуировочной зависимости определяли для каждого элемента концентрации, соответствующие величинам абсорбции анализируемой пробы. Для оценки отклонения находили центр распределения выборки, используя среднее
где Отклонение от среднего в - разность между единичным результатом и средним без учета знака:
Среднее отклонение
Дисперсия и стандартное отклонение характеризуют рассеяние вариант относительно среднего. Дисперсию выборки (V), содержащей n вариант вычисляли по формуле:
Стандартное отклонение (s) представляет собой квадратный корень из дисперсии, взятый с положительным знаком, и имеет размерность измеряемой величины:
Для определения истинного значения в выборочной совокупности использовали следующую формулу:
где s - стандартное отклонение выборки; tp - коэффициент Стьюдента; принимали равным 0,95. Число степеней свободы f = n – 1. При P = 95%, f = 5 - 1 = 4, tp = 2,78 [8]. Относительное стандартное отклонение (sr ) вычисляли по формуле:
Дисперсия, стандартное отклонение и относительное стандартное отклонение характеризуют воспроизводимость результатов химического анализа. Коэффициент чувствительности (S) характеризует отклик аналитического сигнала на содержание компонента y = Sx. Коэффициент чувствительности – это значение первой производной градуировочной функции при данном определенном содержании. Для прямолинейных градуировочных графиков – это тангенс угла наклона прямой:
Чем больше коэффициент чувствительности S, тем меньшие количества компонента можно обнаруживать и определять, получая один и тот же аналитический сигнал. Чем выше S, тем точнее можно определить одно и то же количество вещества. Нижняя граница определяемых концентраций – это наименьшее содержание компонента, определяемого по данной методике.
где s – стандартное отклонение; c - концентрация, 3. ОБРАБОТКА РЕЗУЛЬТАТОВ
Количественное определение примесей в образцах марганца марки Мн 998 проводили методом атомно-абсорбционной спектрометрии на приборе GBC “AVANTA PM” (Австралия). Для этого навеску металлического марганца марки Мн 998 10,000 г. поместили в термостойкую стеклянную коническую колбу вместимостью 200 см3 , растворили в 50 см3 раствора 30% азотной кислоты (1:1 по объему) при нагревании, затем раствор прокипятили до удаления оксида азота. Охлажденный раствор перенесли в мерную колбу вместимостью 500 см3 , довили бидистиллированной водой до метки и перемешали. Расчет систематической ошибки приготовления раствора марганца показал, что данная величина очень мала, поэтому ей можно пренебречь при определении погрешностей результатов анализа металлических примесей в цинке. По ГОСТам определили что в марганце могут содержаться такие металлические примеси как: железо, никель, медь, кальций, магний, кремний, алюминий, титан. Наличие в пробе таких примесей как кальций, магний и титан проверили качественно, и получили отрицательный результат. А такие примеси как железо, никель, алюминий и медь определяли количественно. Для количественного определения примесей в образцах марганца были приготовлены серии градуировочных растворов из государственных стандартных образцов (ГСО) состава водного раствора ионов металлов методом объемного разбавления. Растворы из стандартных образцов готовили и использовали при температуре окружающей среды 20˚С. Растворы, приготовленные из водных растворов ГСО, с массовой концентрацией ионов металла 10 мг/дм3 и менее, длительному хранению не подлежат, и в соответствии с рекомендациями их использовали в день приготовления. ГСО хранятся в упакованном виде, вскрытые ампулы государственных стандартных образцов хранению не подлежат. В пламя горелки распыляли холостой раствор (бидистиллированную воду) и серию градуировочных растворов (в порядке увеличения концентрации). После каждого измерения распылительную систему промывали бидистиллированной водой [7]. Обработка результатов измерений абсорбции проводилась программным обеспечением компьютера, находившегося в составе атомно-абсорбционного спектрометра, в автоматическом режиме. На основании данных эксперимента были построены графики зависимости абсорбции от массовой концентрации металла. Затем проводили расчет параметров линейного уравнения градуировочной зависимости и их доверительных интервалов по формулам, представленным в работах выше. По градуировочной зависимости определяли для каждого элемента концентрации, соответствующие величинам абсорбции анализируемой пробы. Далее проводили статистическую обработку результатов по формулам, представленным выше. 3.1 Качественное определение металлических примесей в марганце марки Мн 998
Для количественного определения металлических примесей в марганце марки Мн 998 анализируемый раствор распыляли в пламени горелки и измеряли абсорбцию, предварительно выбирая лампы с длинами волн подходящими для анализируемой примеси. В результате эксперимента прибор показал значения абсорбции для титана, кальция и магния равные нулю. Из чего мы сделали вывод, что дальнейшее количественное определение данных примесей бесполезно, так как их концентрация в пробе ниже нижней границы определяемых концентраций. А для примесей алюминия, железа, меди и никеля значения абсорбции были отличны от нуля, из чего был сделан вывод продолжении работы.
3.2 Построение градуировочного графика для алюминия
В таблице №8 представлены данные для построения градуировочного графика для алюминия: Таблица №8 Данные для построения градуировочного графика для алюминия.
3.3 Статистическая обработка графика градуировочной зависимости для алюминия
Для оценки параметров линейности полученной градуировочной зависимости мы провели статистическую обработку результатов по формулам представленным выше. Расчет проводили как с использованием формул, так и с помощью программы Microsoft Excel. Результаты представлены в таблицах №9, №10. Таблица №9. Статистические данные.
Таблица №10. Результаты обработки графика градуировочной зависимости для алюминия.
Коэффициент корреляции – R = 0,999968281. 3.4 Построение градуировочного графика для железа
В таблице №11 представлены данные для построения градуировочного графика для железа. Таблица №11. Данные для построения градуировочного графика для железа.
3.5 Статистическая обработка графика градуировочной зависимости для железа
Для оценки параметров линейности полученной градуировочной зависимости мы провели статистическую обработку результатов по формулам представленным выше. Расчет проводили как с использованием формул, так и с помощью программы Microsoft Excel. Результаты представлены в таблицах №12, №13.
Таблица №12. Статистические данные.
Таблица №13. Результаты обработки графика градуировочной зависимости для железа.
Коэффициент корреляции – R = 0,999965406. 3.6 Построение градуировочной зависимости для меди
В таблице №14 представлены данные для построения градуировочной зависимости для меди. Таблица №14. Данные для построения градуировочного графика для меди.
3.7 Статистическая обработка графика градуировочной зависимости для меди
Для оценки параметров линейности полученной градуировочной зависимости мы провели статистическую обработку результатов по формулам представленным выше. Расчет проводили как с использованием формул, так и с помощью программы Microsoft Excel. Результаты представлены в таблицах №15, №16.
Таблица №15. Статистические данные.
Таблица №16. Результаты обработки графика градуировочной зависимости для меди.
Коэффициент корреляции – R = 0,999819876. 3.8 Построение градуировочной зависимости для никеля
В таблице №17 представлены данные для построения градуировочной зависимости для никеля: точно известная концентрация никеля в стандартном растворе и полученная абсорбция. Таблица №17. Данные для построения градуировочного графика для никеля.
3.9 Статистическая обработка графика градуировочной зависимости для никеля
Для оценки параметров линейности полученной градуировочной зависимости мы провели статистическую обработку результатов по формулам представленным выше. Расчет проводили как с использованием формул, так и с помощью программы Microsoft Excel. Результаты представлены в таблицах №18, №19.
Таблица №18. Статистические данные.
Таблица №19. Результаты обработки графика градуировочной зависимости для никеля.
Коэффициент корреляции – R = 0,999869183. 3.10 Количественное определение алюминия в образцах марганца марки Мн 998
Для проведения количественного анализа содержания алюминия в образцах марганца марки Мн 998 было отобрано 5 проб порошкообразного марганца. Результаты представлены в таблице №20. Таблица №20. Анализ образцов марганца марки Мн 998 на содержание алюминия.
Была проведена статистическая обработка полученных результатов для оценки воспроизводимости. Полученные результаты представлены в таблицах №21 и №22. Таблица №21. Статистические данные для количественного определения аллюминия в образцах марганца марки Мн 988.
Таблица №22. Сравнительная характеристика экспериментальных и представленных в ГОСТ 16698.10-71 погрешностей результата анализа алюминия относительных стандартных отклонений.
Таблица №22. Значения коэффициента чувствительности и нижней границы определяемых концентраций для алюминия.
3.11 Количественное определение железа в образцах марганца марки Мн 998
Для проведения количественного анализа содержания железа в образцах марганца марки Мн 998 было отобрано 5 проб порошкообразного цинка. Результаты представлены в таблице №23. Таблица №23. Анализ образцов марганца марки Мн 998 на содержание железа.
Была проведена статистическая обработка полученных результатов для оценки воспроизводимости. Полученные результаты представлены в таблицах №24, №25, №26. Таблица №24. Статистические данные для количественного определения железа в образцах марганца марки Мн 988.
Таблица №25. Сравнительная характеристика экспериментальных и представленных в ГОСТ 16698.6-93 погрешностей результата анализа железа и относительных стандартных отклонений.
Таблица №26. Значения коэффициента чувствительности и нижней границы определяемых концентраций для железа.
3.12 Количественное определение меди в образцах марганца марки Мн 998
Для проведения количественного анализа содержания меди в образцах марганца марки Мн 998 было отобрано 5 проб порошкообразного цинка. Результаты представлены в таблице №27. Таблица №27. Анализ образцов марганца марки Мн 998 на содержание меди.
Была проведена статистическая обработка полученных результатов для оценки воспроизводимости. Полученные результаты представлены в таблицах №28, №29, №30. Таблица №28. Статистические данные для количественного определения меди в образцах марганца марки Мн 988.
Таблица №29. Сравнительная характеристика экспериментальных и представленных в ГОСТ 16698.9-71 погрешностей результата анализа меди и относительных стандартных отклонений.
Таблица №30. Значения коэффициента чувствительности и нижней границы определяемых концентраций для меди.
3.13 Количественное определение никеля в образцах марганца марки Мн 998
Для проведения количественного анализа содержания никеля в образцах марганца марки Мн 998 было отобрано 5 проб порошкообразного цинка. Результаты представлены в таблице №31. Таблица №31 Анализ образцов марганца марки Мн 998 на содержание никеля.
Была проведена статистическая обработка полученных результатов для оценки воспроизводимости. Полученные результаты представлены в таблицах №32, №33, №34. Таблица №32. Статистические данные для количественного определения никеля в образцах марганца марки Мн 988.
Таблица №33. Сравнительная характеристика экспериментальных и представленных в ГОСТ 16698.7-71 погрешностей результата анализа никеля и относительных стандартных отклонений.
Таблица №34. Значения коэффициента чувствительности и нижней границы определяемых концентраций для никеля.
3.14 Количественная характеристика примесей алюминия, железа, меди, никеля в образцах марганца марки Мн 998
Полученные результаты свидетельствуют, что в исследуемых пробах марганца содержатся такие металлы, как алюминий, железо, медь и никель. Установлено, что кальций, магний и титан отсутствуют. Результаты представлены в таблице №35. Таблица №35. Количественная характеристика примесей алюминия, железа, меди, никеля, кальций, магния и титана в образцах марганца марки Мн 998.
* Массовая доля элемента ниже нижней границы определяемых концентраций. 3.15 Значения относительных стандартных отклонений, коэффициентов чувствительности и нижних границ определяемых концентраций для металлических примесей обнаруженных в составе марганца марки Мн 998
В таблице №36 представлены результаты статистической обработки результатов эксперимента. Таблица №36.
Статистическая обработка выборок экспериментальных данных дала узкие доверительные интервалы, что свидетельствует о хорошей воспроизводимости применяемых методик. Малые величины дисперсий, стандартных отклонений и относительных стандартных отклонений также подтверждают хорошую воспроизводимость методик атомно-абсорбционного анализа.
ВЫВОДЫ
1. Проведенный анализ доказал что метод атомно-абсорбционной спектрометрии является наиболее подходящим для проведения анализа марганца. 2. На приборе GBC “AVANTA PM” (Австралия) был проведен анализ примесей алюминия, железа, меди, титана, кальция, магния и никеля в образцах марганца марки Мн 998. Полученные результаты свидетельствуют, что в исследуемых пробах марганца содержатся такие примеси, как железо, алюминий, медь и никель. Так же установлено, что концентрации титана, кальция и магния ниже коэффициента чувствительности прибора, поэтому можно сделать вывод, что данные примеси в пробе отсутствуют. 3. Статистическая обработка выборок экспериментальных данных дала узкие доверительные интервалы, что подтверждает хорошую воспроизводимость используемых в анализе методик количественного определения алюминия, железа, меди и никеля. Малые величины дисперсий, стандартных отклонений и относительных стандартных отклонений также свидетельствуют о хорошей воспроизводимости данных методик атомно-абсорбционного анализа. 4. Найденное содержание металлических примесей хорошо согласуется с результатами, представленными в ГОСТе 6008 – 90 на марганец марки Мн 998.
СПИСОК ЛИТЕРАТУРЫ
1. Составители Крысанова Т. А., Котова Д. Л., Бабенко Н. К. Атомно абсорбционная спектрометрия, учебно-методическое пособие по специальностям химия, биология. - В.: Химия, 2005. – 31 С. 2. Ермаченко Л. А. Атомно абсорбционный анализ в санитарно гигиенических исследованиях. – М.: Химия, 1997. – 274 С. 3. Крешков А.П. Основы аналитической химии. – М.: Химия, 1976. – Т.1. Теоретические основы. – 472 С., – Т.2. Теоретические основы. Количественный анализ. – 480 С., – Т.3. Физико-химические (инструментальные) методы анализа. – 472 С. 4. Васильев В.П. Аналитическая химия. Физико – химические методы анализа. – М.: Высш. шк., 2003. – Т.2. – 384 С. 5. Отто М. Современные методы аналитической химии. – М.: Техносфера, 2003. – Т.1. – 416 С. 6. Глубоков Ю.М., Головачева В.А., Ефимова Ю.А. Аналитическая химия. / Под ред. А.А. Ищенко – М.: Академия, 2004. – 320 С. 7. Ермаченко Л.А. Атомно-абсорбционный анализ в санитарно – гигиенических исследованиях. / Под ред. Л.Г. Подуновой – М.: Чувашия, 1997. – 208 С. 8. Золотов Ю.А. Основы аналитической химии. – М.: Высш. шк., 2002. – Т.1. Общие вопросы. Методы разделения. – 351 с. – Т.2. Методы химического анализа. – 494 С. 9. Рекламный сайт кампании «Люмэкс». – С.-П., Россия. http://www.lumex.ru/equipment.php?id=33. 10. ГОСТ 1770–74 (ИСО 1042-83, ИСО 4788-80). Посуда мерная лабораторная стеклянная. Цилиндры, мензурки, колбы, пробирки. Общие технические условия. 11. Дорохова Е.Н., Прохорова Г.В. Задачи и вопросы по аналитической химии. – М.: Мир, 2001. – 267 С. 12. Инструкция к прибору GBC “AVANTA PM” (Австралия) по использованию методов атомной абсорбции в пламени. – 29 С. 13. РД 52.24.377–95. Методические указания. Атомно-абсорбционное определение металлов (Al, Ag, Be, Cd, Co, Cr, Cu, Fe, Mn, Mo, Ni, Pb, V, Zn) в поверхностных водах суши с прямой электротермической атомизацией проб, утвержденные Росгидрометом. 14. Инструкция по применению государственных стандартных образцов состава водных растворов ионов алюминия ГСО 8059-2004. – 1 С. 15. Инструкция по применению государственных стандартных образцов состава водных растворов ионов железа (III) ГСО 7766-2006. – 1 С. 16. Инструкция по применению государственных стандартных образцов состава водных растворов ионов меди ГСО 7764-2006. – 1 С. 17. Инструкция по применению государственных стандартных образцов состава водных растворов ионов никеля ГСО 7785-2006. – 1 С. 18. ГОСТ 16698.4-93. Фотометрические методы определения фосфора в металлическом и азотированном марганце. – М.: Госстандарт России, 1995. – 10 С. 19. ГОСТ 16698.5-71. Методы определения кремния в металлическом и азотированном марганце. – М.: Госстандарт России, 1995. – 10 С. 20. ГОСТ 16698.6-71. Методы определения железа в металлическом и азотированном марганце. – М.: Госстандарт России, 1995. – 10 С. 21. ГОСТ 16698.7-71. Фотометрический и атомно-абсорбционный методы определения никеля в металлическом марганце и металлическом азотированном марганце. – М.: Госстандарт России, 1998. – 10 С. 22. ГОСТ 16698.9-71. Фотометрический и атомно-абсорбционный методы определения меди в металлическом марганце и металлическом азотированном марганце. – М.: Госстандарт России, 1997. – 10 С. 23. ГОСТ 16698.12-84. Атомно-абсорбционный метод определения кальция и магния в металлическом марганце и металлическом азотированном марганце. – М.: Госстандарт России, 1999. – 10 С. 24. ГОСТ 16698.10-71. Фотометрический метод определения алюминия в металлическом марганце и металлическом азотированном марганце. – М.: Госстандарт России, 1997. – 10 С. 25. ГОСТ 16698.11-71. Фотометрический метод определения титана в металлическом марганце и металлическом азотированном марганце. – М.: Госстандарт России, 1995. – 10 С. 26. ГОСТ 16698.1-93. Определения марганца в металлическом и азотированном марганце. – М.: Госстандарт России, 1995. – 10 С. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


 (1)
(1)
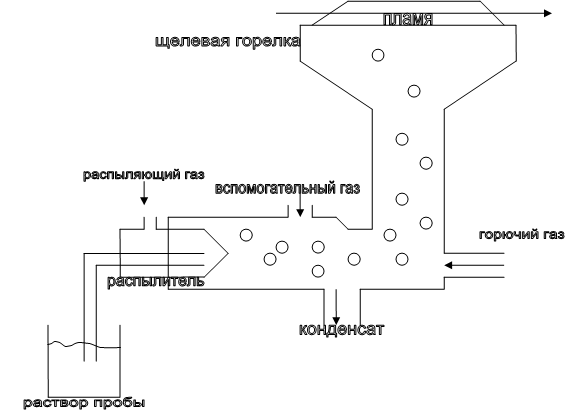



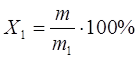
 ,
,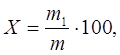



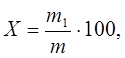




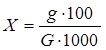
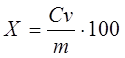
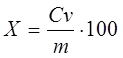


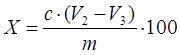
 ,
,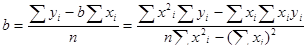 .
.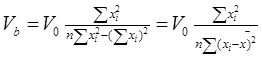 ,
,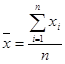 .
. ,
, .
.