Поглощение света
Контрольная работа
Поглощение света
Содержание
1. Закон Бугера-Ламберта-Бера
2. Спектры поглощения света
3. Основы спектрофотометрии
4. Спектры поглощения белков
5. Спектры поглощения нуклеиновых кислот
6. Некоторые факторы, влияющие на адсорбционные свойства хромофоров
7. Несколько примеров применения абсорбционной спектроскопии
Литература
1. Закон Бугера-Ламберта-Бера
Способность вещества поглощать свет зависит от ряда факторов: электронного строения атомов и молекул, концентрации поглощающих центров, толщины поглощающего слоя и т.д. Впервые этот эффект был изучен Пьером Бугером в 1729 г., который определил количество света, теряющегося при прохождении определенного пути в атмосфере. Как экспериментально установлено Иоганом Ламбертом (1760), интенсивность света, прошедшего через образец, экспоненциально зависит от его толщины:
I(x) = Io exp(-k1 x).
Август Бер (1852) установил аналогичную зависимость от концентрации поглощающих молекул с:
I(с) = Io exp(-k2 с),
где I0 – интенсивность падающего света, а k1 и k2 – коэффициенты пропорциональности. Величина T = I/I0 называется коэффициентом пропускания света, а величина
= Iпогл/I0 = (I0 – I)/I0 = 1-T
коэффициентом поглощения света.
Отсюда можно вывести закон поглощения света в более общем виде. Рассмотрим прохождение монохроматического света через раствор хромофора - окрашенного вещества, т.е. способного поглощать свет, помещенного в прозрачную кювету толщиной l (Рис.1).
I
l
dl
I0
Рис.1. Прохождение света через образец толщиной l. Io и I – интенсивность падающего и прошедшего света, соответственно.
Пусть в 1 см3 находится n поглощающих молекул, т.е. их концентрация равна n (см–3). Тогда ослабление света при прохождении им тонкого слоя dl равно:
dI/I = - s n dl,
где коэффициент пропорциональности s называется поперечным сечением поглощения света молекулой хромофора. Размерность s – см Смысл этой величины заключается в том, что вероятность поглощения фотона при прохождении света через площадку s равна 1. Величина s определяется видом молекулы, строением ее электронных оболочек, энергетическими уровнями внешних (валентных) электронов и зависит от энергии квантов света, т.е. от его длины волны : s= s().
Интегрируя это выражение:
,
получим:
I = I0 e-snl
Это закон Бугера-Ламберта-Бера - важнейший закон фотобиологии.
Вместо числа молекул в 1 см3, удобней использовать молярную концентрацию вещества, с [моль/л], и применять десятичные логарифмы. Тогда закон Бугера-Ламберта-Бера будет выражен в более удобном виде:
I = I0 10-()cl или I = I0 10-D.
Здесь коэффициент пропорциональности () называется молярным коэффициентом поглощения или экстинкцией, а величина D() = ()cl - оптической плотностью (в английской литературе используется обозначение A от английского слова absorbance). Она связана с коэффициентом пропускания света T = I/I0 соотношением:
D = -lg T.
При малых D < 0.05 коэффициент поглощения пропорционален оптической плотности:
= 1-T = 1- 10 D D ln10 0.434 D
2. Спектры поглощения света
Величины () и D(), как и s(), зависят от набора энергетических уровней электронов в молекуле, т.к. поглощается только свет, способный вызвать соответствующие переходы между ними. Поэтому, изучая зависимости () и D(), которые называются спектрами поглощения света, можно получить картину энергетических уровней молекулы. Величина (), в отличие от D(), зависящей от концентрации вещества и толщины кюветы, зависит только от природы данного вещества, строения его электронных оболочек, и поэтому является его индивидуальной характеристикой. Для хорошо поглощающих свет хромофоров характерны величины порядка 103-105 л/мольсм.
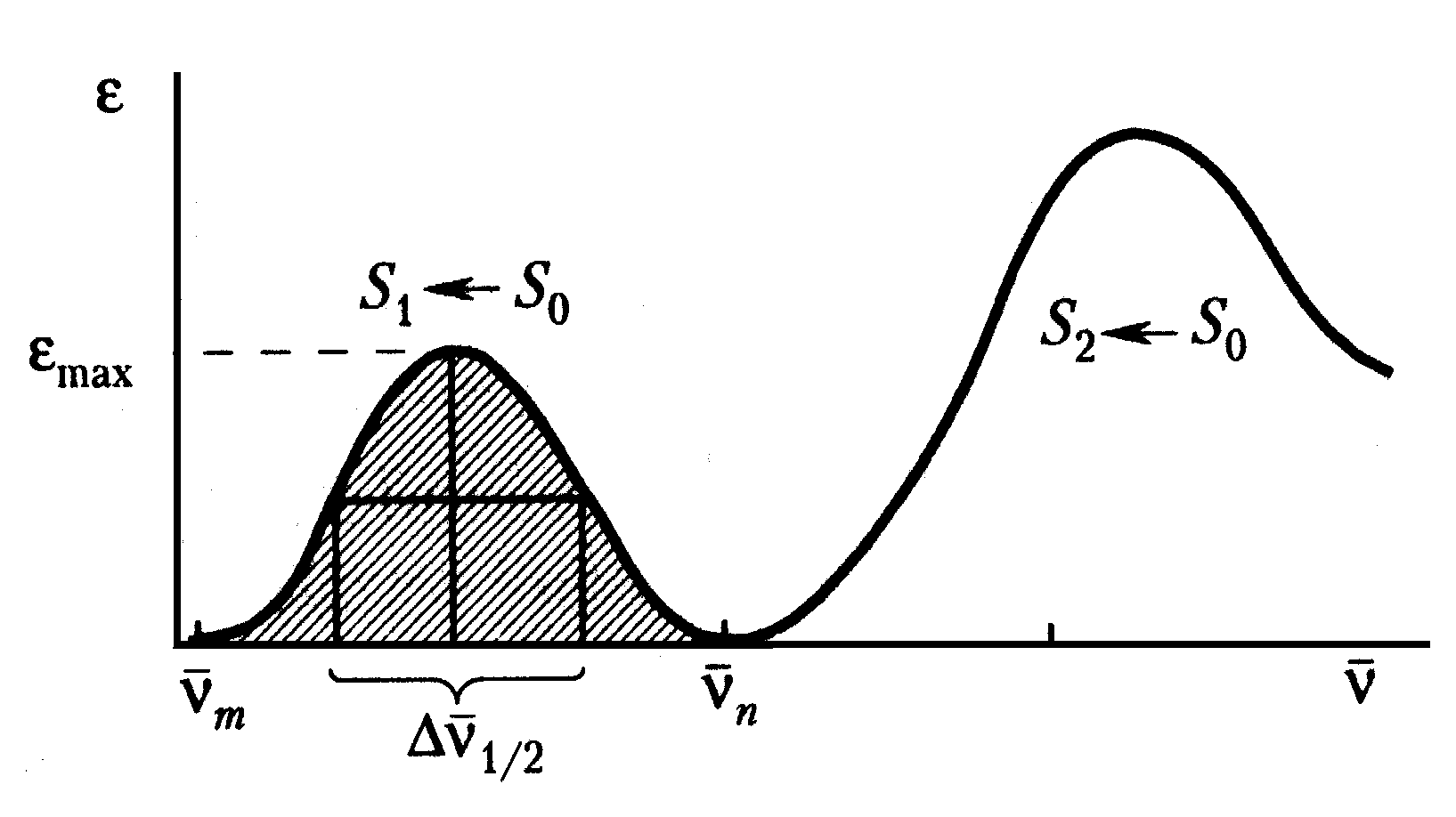
Рис. 2 Полосы поглощения света растворами хромофоров. Вторая полоса может соответствовать переходу на второй возбужденный уровень или поглощению света другой хромофорной группой (по Владимирову и Потапенко, 2006).
Спектры поглощения атомов в газообразном состоянии, где они практически не взаимодействуют, линейчатые и точно соответствуют дискретным энергетическим уровням атомов. В сложных органических молекулах или в конденсированных средах, например, в растворах, спектры поглощения света () имеют квазинепрерывный вид и представляют собой кривые Гауссового вида (Рис.2) в шкале частот (или энергий фотонов). Уширение спектральных линий происходит вследствие расщепления электронного уровня на множество колебательно-вращательных подуровней. Кроме того, в конденсированных средах, например, в растворах, молекулы хромофора, окружены полярными молекулами растворителя. Их дипольные моменты формируют локальные электрические поля, влияющие на энергии электронных переходов, а следовательно и на положение спектральных линий. Они не квантуются, и это позволяет получить непрерывный набор подсостояний. Суперпозиция большого числа возможных состояний и обусловливает гауссову форму полосы поглощения с центром в точке max, соответствующей частоте наиболее вероятного электронного перехода.
Спектры поглощения света органическими молекулами могут содержать одну или несколько полос, имеющих вид гауссовых кривых в энергетической шкале () или (Рис.2). Они характеризуются:
- положением максимумов, т.е. величинами max или max, соответствующими энергиями наиболее вероятных электронных переходов;
- числом максимумов, зависящим от числа хромофорных групп в молекуле и взаимодействия их со средой. Например, в спектре поглощения хлорофилла (Рис.3Д) выделяются две полосы в синей и красной области спектра, соответствующие поглощению света порфириновым кольцом и атомом магния, координированным в центре этого кольца. В зависимости от микроокружения магния, красная полоса может существенно смещаться на десятки нанометров. В спектре поглощения хромофора иногда присутствует коротковолновая полоса (Рис.2), соответствующая переходу на второй возбужденный уровень (S0 S2), если энергия такого перехода не слишком велика и соответствует ультрафиолетовой области спектра (>200 нм), которую еще можно изучать с помощью оптических приборов1;
- полушириной 1/2, соответствующей ширине полосы на половине ее высоты: 1/2max (Рис.2). Она зависит от энергий колебательных и вращательных компонентов, от взаимодействия со средой и обратно пропорциональна времени жизни возбужденного состояния: 1/2 1/,
- величиной максимума max, примерно пропорциональной площади под спектральной кривой (Рис.2): , которая пропорциональна силе осциллятора fmn, характеризующей вероятность электронного перехода:
.
Действительно, для кривых нормального распределения (гауссовых кривых) существует приближенное соотношение между величиной максимума, площадью под кривой и полушириной:
Очень важную роль в поглощении света биомолекулами играют углеводородные цепочки с сопряженными связями, в которых чередуются одинарные и двойные связи. В них происходит обобществление -электронов, которые делокализуются, т.е. распределяются на всю длину цепочки. Это приводит к снижению их общего энергетического уровня до нескольких электрон-вольт - энергии, характерной для ультрафиолетового и видимого диапазонов. Чем длиннее цепочка, т.е. чем выше степень делокализации электронов, тем дальше в красную область спектра смещаются спектры поглощения таких молекул.
Владимиров и Потапенко (2007) приводят простой расчет, качественно доказывающий это утверждение. Если взять цепочку из N звеньев длиной l и общей длиной L=Nl, в которой каждое звено образовано одной одинарной и одной двойной связью, и предположить, что энергия -электронов в этой цепочке сопряженных связей характеризуется прямоугольной потенциальной ямой, то можно определить распределение этих электронов по энергиям. Действительно, согласно Де Бройлю, движение электрона со скоростью v может быть описано как распространение волны с длиной:
=h/mev ,
где h – постоянная Планка. Электрон и характеризующая его волна не должны выходить за пределы цепочки. При этом устойчивое состояние такой системы наблюдается, когда внутри потенциальной ямы возникают стоячие волны с узлами на стенках, т.е. в яме должно помещаться целое число полуволн:
L=n/2,
Где n=1,2,3... Следовательно, скорость электрона может принимать дискретные значения:
v = h/me =n h/2meL,
а его кинетическая энергия также будет квантована:
En = mev2/2=n2h2/8meL2
Поскольку на каждом энергетическом уровне могут располагаться только два электрона с противоположными спинами, то в цепи из N звеньев помещаются 2N электронов на уровнях с энергиями от h2/8meL2 до N2h2/8meL
Наиболее длинноволновая полоса поглощения света такой цепочкой соответствует электронному переходу с верхней заполненной молекулярной орбитали на нижнюю свободную орбиталь:
h = EN+1 - EN .
Следовательно,
.
Так как L=Nl, то:
.
При N>2:
,
То есть длина волны поглощаемого света пропорциональна N. Действительно, цепочки с двумя сопряженными двойными связями имеют максимум поглощения при 233 нм, с тремя – при 260-280 нм, а с шестью, как у зрительного пигмента ретиналя, – при 360 нм. Это позволяет нам улавливать видимый свет и подвергаться действию ультрафиолета.
На рис.6 приведены спектры поглощения света некоторых важнейших биологических молекул.
3. Основы спектрофотометрии
По спектрам поглощения света можно:
- Идентифицировать вещество по количеству полос, их положению, соотношению максимумов;
- Определить положение возбужденных уровней, т.е. энергии электронных переходов в молекуле хромофора;
- Измерить концентрацию вещества. Поскольку D = c l, то для вещества с известным значением молярного коэффициента поглощения max измерение оптической плотности D на длине волны max в кювете с заданной толщиной l, позволяет сразу определить концентрацию хромофора c. Если же молярная экстинкция max заранее не известна, то сначала строят калибровочный график D(c) для разных концентраций изучаемого вещества и по нему находят величину c для измеренной величины D.
- Изучить динамику химической реакции, измеряя изменение концентрации какого-то из ее компонентов во времени.
На этом построено огромное множество методов аналитической химии и биохимии. Если в изучаемой реакции образуется или, наоборот, исчезает окрашенный, т.е. поглощающий свет продукт, то с помощью спектрофотометра можно определить его концентрацию и проследить за динамикой реакции. Если исследуемое вещество не окрашено, то применяют специальные красители-индикаторы, меняющие поглощение света при реакции с изучаемым веществом. Если, например, нужно изучить реакцию:
А + В С,
где вещества А, В и С не поглощают видимый свет, но в результате взаимодействия одного из них, например, вещества В с индикатором D образуется вещество S, поглощающее свет:
- В + D S,
то измеряя его оптическую плотность, можно определить концентрации веществ А, В и С, которые линейно связаны с D и S.
В спектрофотометре - приборе для регистрации спектров поглощения вещества - фотоприемником непосредственно измеряется интенсивность света I, прошедшего через образец. Сравнивая ее с интенсивностью света, прошедшего через кювету с растворителем (I0), можно определить коэффициенты пропускания и поглощения света: T = I/Io и = 1 – T. Однако, удобно измерять не пропускание Т, а оптическую плотность D, которая, в отличие от Т, является аддитивной величиной: оптическая плотность D смеси из нескольких хромофоров, т.е. поглощающих свет веществ, равна сумме оптических плотностей отдельных хромофоров Di:
D = Di
Рис. 3. Спектры поглощения света важнейших биомолекул. А. ДНК (1 – двухцепочечная ДНК; 2 – ДНК, денатурированная нагреванием; 3 – смесь нуклеотидов. Б – белок (сывороточный альбумин человека. В – зрительный белок родопсин. Г – никотинамидадениндинуклеотид, окисленная и восстановленная формы: NAD+ и NADH. Д – раствор хлорофилла в эфире (1 – хлорофилл а; 2 – хлорофилл b. Е – флавинадениндинуклеотид (флавинаденинмононуклеотид), окисленная и восстановленная формы: FAD (FMN) и FADH2 (FMNH2). Ж – гем в составе цитохрома с, окисленная и восстановленная формы; показаны основные полосы: , и .
Поэтому, вычитая оптическую плотность кюветы с растворителем из общей оптической плотности раствора, можно определить оптическую плотность самого хромофора. Для этого в спектрофотометрах имеется логарифмический блок, преобразующий фототок приемника света, пропорциональный I, в сигнал пропорциональный D:
y = k lg I = K D,
В старых спектрофотометрах эту функцию выполняло специальное электронное устройство, а сейчас ее решают компьютерными средствами.
Приборы для изучения спектров поглощения света называются спектрофотометрами. Они подразделяются на однолучевые и двухлучевые. В однолучевом спектрофотометре (Рис.4А) свет от ксеноновой лампы, излучающей, в отличие от ламп накаливания, не только видимый, но и ультрафиолетовый свет, проходит через монохроматор, который разлагает свет в спектр, из которого с помощью узкой щели выделяет спектральную полосу шириной порядка одного или нескольких нанометров. Для этого используются диспергирующие элементы – кварцевые призмы или, в современных приборах, дифракционные решетки, разлагающие белый свет в спектр. Дифракционные решетки отличаются линейной зависимостью угла отклонения луча и независимостью линейной дисперсии от длины волны, большей светосилой и большей обратной линейной дисперсией (0,1-1,0 нм/мм) по сравнению с призмами (1-10 нм/мм), и, наконец, они дешевле. Однако, в ультрафиолетовой области у них более высокий уровень рассеянного света. Поэтому в ультрафиолетовом диапазоне иногда оправдано применение призменных монохроматоров.
Рис. 4. Схема однолучевого (А), двухлучевого (Б) и двухволнового (В) спектрофотометров. Л – лампа; М – монохроматор; К – кюветы; У – усилитель; Ком – компьютер; П – принтер. (По: Владимиров, Потапенко, 2006).
|
А |
|
|
Б |
|

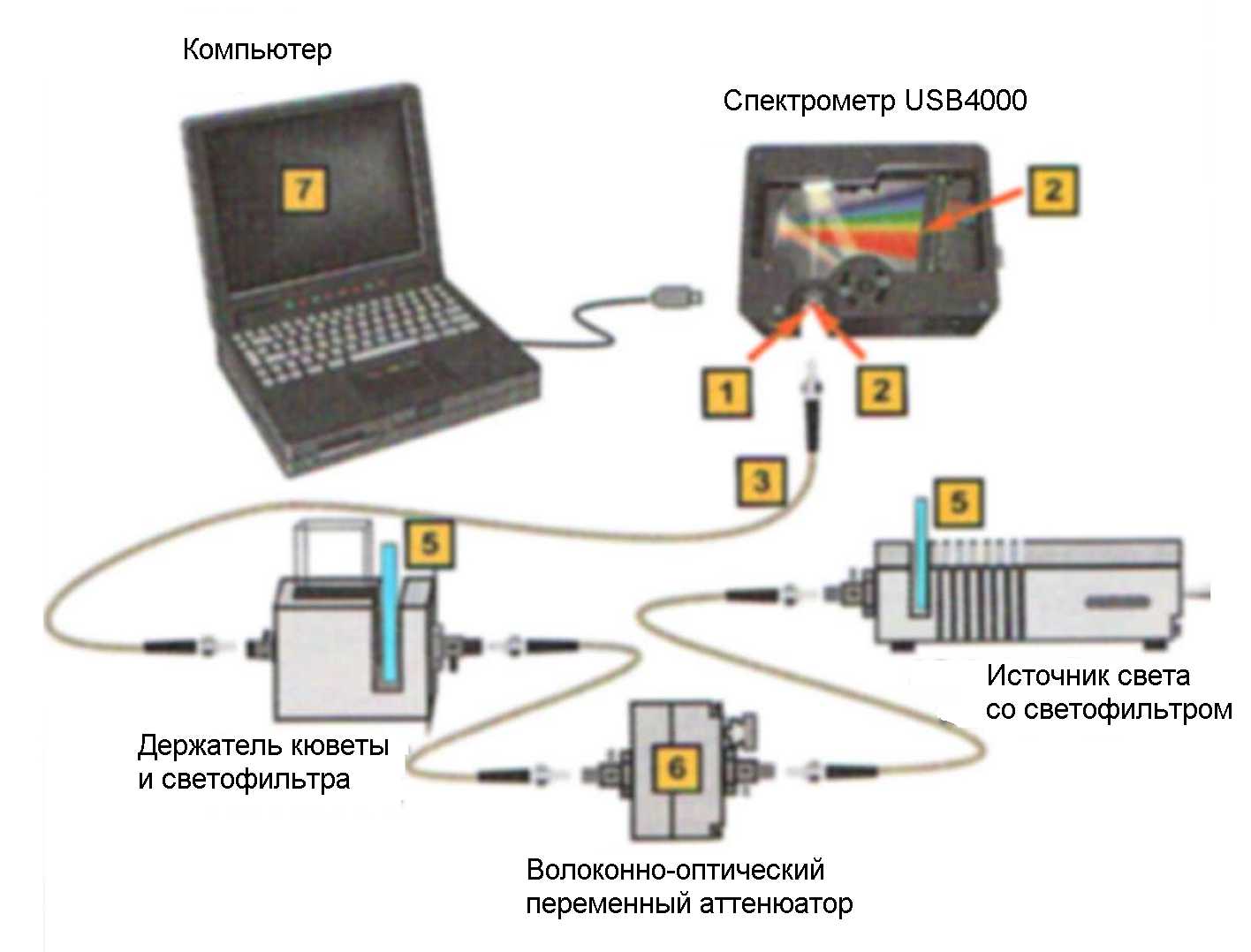
Рис.5. Устройство спектрофотометра USB4000 фирмы Ocean Optics. А – универсальный спектрометр. Б – схема измерений. 1 – коннектор, для подключения оптического волокна; 2 – входная щель шириной от 5 до 200 мкм, регулирующая интенсивность входящего светового потока; 3 – длинноволновой светофильтр, отсекающий коротоковолновое излучение, если оно не нужно для измерений (в других случаях он убирается); 4 – коллимирующее вогнутое зеркало, формирующее параллельный световой пучок; 5 – дифракционная решетка, разлагающая свет в спектр; 6 – фокусирующее зеркало; 7 – собирающая цилиндрическая линза, фокусирующая свет на линейный детектор; 8 – линейка из 3648 CCD элементов, каждый из которых (пиксел) регистрирует свет с определенной длиной волны.
Для более точного выделения монохроматического света с определенной длиной волны применяют двойные монохроматоры, в которых выделенный узкополосный свет дополнительно разлагается второй призмой или решеткой. Если требуется изучить тонкую структуру спектра с максимальным спектральным разрешением, то устанавливается минимальная ширина щели, обычно 0,5-1 нм. Но при этом уменьшается световой поток и требуется максимальная чувствительность фотоприемника. Если не требуется такое высокое спектральное разрешение, то ширину щели увеличивают до 2-5 нм, что существенно снижает требования к чувствительности фотоприемника. Фотоприемник регистрирует свет, прошедший через кювету с образцом. Он, преобразуют световую энергию в электрический ток, пропорциональный интенсивности проходящего света. Далее происходит усиление электрического сигнала, логарифмическое преобразование и регистрация с помощью самописца или компьютера.
Фотоприемником обычно служит фотоэлектронный умножитель (ФЭУ) или в некоторых современных приборах линейка фотодиодов или CCD элементов2 (Рис. 5А) ФЭУ - наиболее чувствительный фотоприемник, способный регистрировать даже единичные фотоны, но он значительно дороже и требует высоковольтного питания (700-3000 В), что усложняет и удорожает прибор. Используя линейку CCD элементов, каждый из которых настроен на определенную длину волны, можно сразу зарегистрировать весь спектр и обойтись без дорогостоящей механики, управляющей движением щели, выделяющей монохроматический свет.
При спектрофотометрических измерениях сначала регистрируют фототок, прошедший через контрольную кювету с растворителем. Эти показания преобразуются в величину, пропорциональную оптической плотности D0, и запоминаются компьютером. Потом регистрируется фототок, прошедший через ту же кювету, в которую добавлен исследуемый раствор. После логарифмического преобразования сигнала и вычитания D0 находят чистую оптическую плотность добавленного вещества: Dв = D1 – D0. Достоинства однолучевого спектрофотометра – простота и дешевизна, а недостатки - нестабильность излучения лампы, шумы приемника света, что снижает чувствительность прибора. Обычно чувствительность однолучевых спектрофотометров порядка 10-3 D.
В качестве примера современного спектрофотометра приведем прибор USB4000 фирмы Ocean Optics (Рис.5). В нем используется многоцелевой волоконно-оптический спектрометр с дифракционной решеткой и линейкой из 3648 CCD элементов. Особенность прибора в том, что свет разлагается в спектр не до, а после прохождения образца. Это позволяет использовать спектрометр как дешевый и миниатюрный (размером всего 15х5 см) универсальный модуль, пригодный как для спектрофотометрических, так и для спектрофлуорометрических измерений. Волоконная оптика позволяет измерять спектры удаленных образцов.
Более чувствительны дифференциальные спектрофотометры, в которых регистрируется не абсолютная величина оптической плотности, а разница в оптической плотности или двух образцов или одного и того же образца при некотором изменении спектра, вызванном химической модификацией, воздействием внешнего фактора или функционированием данной молекулы. В двухлучевом спектрофотометре (Рис.4Б) луч от лампы после монохроматора расщепляется на два с помощью обтюратора - вращающегося зеркала, расположенного под углом 45о относительно направления луча. Это зеркало имеет такие вырезы, что половину времени монохроматический луч проходит прямо через них на первый образец, а вторую половину отражается зеркалом на другое зеркало и идет через второй образец. Фотоприемник попеременно улавливает свет, прошедший то через одну кювету, то через вторую. Оптические плотности, измеренные в каждом канале, сравниваются, и регистрируется их разница: D = D1 – D Этот метод позволяет измерять малые изменения оптической плотности до 10-4 D на фоне общего большого поглощения. Его недостаток - сложность и более высокая стоимость прибора.
Рис. 6. Схема измерения молярного коэффициента поглощения света двухволновым методом
Двухволновой спектрофотометр (Рис.4В) используется для изучения образцов, сильно рассеивающих свет, например, клеточных суспензий. Рэлеевское светорассеяние сильно возрастает в ультрафиолетовой области и его вклад в регистрируемый свет (отрезок 2+3 на Рис.6) становится сравнимым с вкладом поглощения света (отрезок 1). Для того, чтобы вычесть эту компоненту, в двухволновом спектрофотометре свет от лампы расщепляется на два луча и проходит через два монохроматора, выделяющих две близкие длины волны 1 и 2, одна из которых соответствует изучаемому максимуму, а вторая расположена у его подножия. На волне 1 мы регистрируем сумму поглощения света (отрезок 1 на Рис.6) и рассеяния (отрезки 2+3), а на волне 2 - только рассеяние (3). Величина светорассеяния в этих близких точках различается на сравнительно небольшую величину (отрезок 2 на рис. 6). Поэтому вычитание интенсивности света, регистрируемого в одном канале, от интенсивности света в другом канале позволяет почти полностью исключить эту компоненту:
I(1) – I(2) = Iпогл(1) + Iрасс(1) - Iпогл(2) - Iрасс(2) Iпогл(1),
А поскольку при 2 интенсивность поглощенного света невелика: Iпогл(2)0, а рассеяние различается на небольшую величину: Iрасс(1) Iрасс(2) то мы практически измеряем интенсивность света в максимуме поглощения. Относительная ошибка измерений (2)/(1) значительно уменьшилась по сравнению с начальной: (2+3)/(1), где в скобках приведены высоты соответствующих отрезков на Рис.9. Двухволновые спектрофотометры также позволяют снизить помехи, связанные динамическими изменениями светорассеяния вследствие оседания или подвижности биообъектов. Они удобны для кинетических измерений, например, для изучения процессов электронного транспорта в митохондриях или целых клетках. Двухволновой спектрофотометр - очень точный прибор, но сложный и дорогой, так как в нем используются два монохроматора.
Для исследования быстропротекающих фотопроцессов используются методы импульсной спектроскопии. Они были разработаны в 1950-х годах и получили название фдеш-фотолиза. Суть его состоит в исследовании короткоживущих промежуточных фотопродуктов (интермедиатов), образующихся при действии кратковременной мощной вспышки света (flash), с помощью последующего измерения кинетики изменения поглощения света на определенной длине волны, спектров поглощения или флуоресценции этих фотопродуктов через определенный промежуток времени (он может быть очень коротким). Для возбуждения молекул используют импульсные лампы (длительность вспышки порядка микросекунд) или импульсные лазеры, дающие нано-, пико-и даже фемтосекундные (10-8 10-13 с) импульсы. В качестве зондирующего импульса, используемого для регистрации поглощения света, используют более слабую вспышку света или лазерный импульс. Он не должен вызывать фотопревращений биологического объекта, но быть достаточным для надежной регистрации спектральных изменений.
Необходимо помнить, что при D 2 наступает насыщение и прибор не может правильно измерить эту величину. Согласно Владимирову и Потапенко (2006), наименьшая погрешность измерений оптической плотности достигается в интервале 0,2 - 0,8. Поэтому если раствор хромофора чересчур оптически плотен, то его необходимо разбавлять так, чтобы измеряемая величина D попадала в этот интервал. Если величина D недостаточна, то целесообразно или взять кювету с большей толщиной, или увеличить интенсивность падающего света, увеличив ширину щели.
В некоторых случаях закон Бугера-Ламберта-Бера может нарушаться:
- при использовании немонохроматического света зависимость D(с) становится нелинейной;
- при существенном светорассеянии образца, такого как, например, клеточная суспензия, величина оптической плотности D в ультрафиолетовой области будет завышаться, так как измеряемое ослабление света будет состоять не только из поглощения, но и рассеяния;
- при сильной люминесценции образца, испускаемые фотоны могут попадать в фотоприемник, занижая величину D.
- при неравномерном распределении хромофора, например, в виде отдельных гранул, что часто происходит при агрегации молекул, оптическая плотность раствора будет ниже, чем в случае гомогенного распределения (эффект сита).
4. Спектры поглощения белков
Спектры поглощения белков имеют два максимума: при 190 и 280нм (Рис.7). Первый максимум при 190 нм, почти на коротковолновом пределе возможности спектрофотометров, обусловлен поглощением света пептидной группой –CO-NH- . В качестве простой модели пептидной группы, на которой были проведены квантово-механические расчеты, служили амиды R1-CO-NH-R В пептидной или амидной группе все четыре атома N, C, O и Н лежат в одной плоскости (Рис.8А) и вследствие взаимодействия электронных орбиталей -электроны делокализованы по атомам N, C и O, то есть связь N-C на самом деле не одинарная, а связь С=О - не двойная. Они обе носят частично двойной характер. В этой группе есть три -орбиты, две из которых заняты в основном состоянии. Кроме того, есть пара неподеленных электронов, локализованная на атоме кислорода. Электронные переходы * поляризованы в плоскости группы. Такие переходы дают наиболее интенсивные полосы поглощения в молекулярных спектрах благодаря высокой степени перекрывания волновых функций основного и возбужденного состояний. Из них переход 1 соответствует полосе поглощения при 190 нм (Рис.7Б). Переход 2, соответствующий длине волны 165 нм, получен расчетным путем, и зарегистрировать его не удается. Переход n (220 нм) выражен слабо, т.к. электронные облака неподеленной пары n-электронов в атоме кислорода и общих -электронов в возбужденном состоянии очень слабо перекрываются, потому что их орбитали лежат в почти перпендикулярных плоскостях.
Рис.7. Спектры поглощения спиральной и клубкообразной поли-L-глутаминовой кислоты (По: М. Франк-Каменецкий, 1967).
На поглощение света в области 190 нм влияет спиральность молекулы. У полипептида с -спиральной структурой молярный коэффициент поглощения 190 примерно на 40 % меньше, чем у развернутого полипептида. Этот гипохромный эффект обусловлен почти параллельной ориентацией дипольных моментов электронных переходов в пептидных группах соседних витков, которые в –спиральной структуре располагаются почти параллельно, а в клубке – хаотично. При такой пространственной корреляции между ними возникает электростатическое взаимодействие, приводящее к ослаблению поглощения. По гипохромному эффекту можно сравнивать спиральность молекул разных белков. Если принять степень регулярности высокоспирального парамиозина за 100 %, то у миоглобина она будет 83 %, у инсулина - 66 %, у рибонуклеазы - 40 %.
Рис.8. Волновые функции, соответствующие наиболее подвижным электронам в амидной группе (А), и основные электронные переходы (Б) (по: Волькенштейн, 1975).
Максимум при 280 нм обусловлен поглощением -электронной системы ароматических аминокислот триптофана, тирозина и фенилаланина. Наибольшим поглощением на этой длине волны отличается триптофан. Примерно на порядок слабее поглощает тирозин и еще на порядок – фенилаланин: три >> тир >> фен. Поэтому если в белке есть хоть один триптофанил, то общее поглощение на длине волны 280 нм будет определяться этим аминокислотным остатком. Этот максимум значительно, на порядок слабее максимума при 190 нм, потому что количество триптофанилов в белке невелико, единицы, а пептидные группы есть в каждом звене. Тем не менее, поглощение УФ света на длине волны 280 нм характерно для всех белков и позволяет идентифицировать их в сложных смесях веществ и в клетках.
5. Спектры поглощения нуклеиновых кислот
Поглощение света нуклеотидами и нуклеиновыми кислотами обусловлено поглощением света пуриновыми и пиримидиновыми азотистыми основаниями. Пиримидины – тимин, цитозин и урацил - имеют три нижних орбиты, занятых 6 электронами и три незанятых верхних * орбиты, а также две пары неподеленных электронов у атомов азота (n орбиты). У пуринов – аденина и гуанина – есть пять нижних занятых орбит и четыре незанятых верхних * орбит, а также три n орбиты (Рис.9). Поглощение света в обоих случаях практически полностью обусловлено * переходами, которые, в отличие от n* переходов, поляризованы в плоскости оснований.
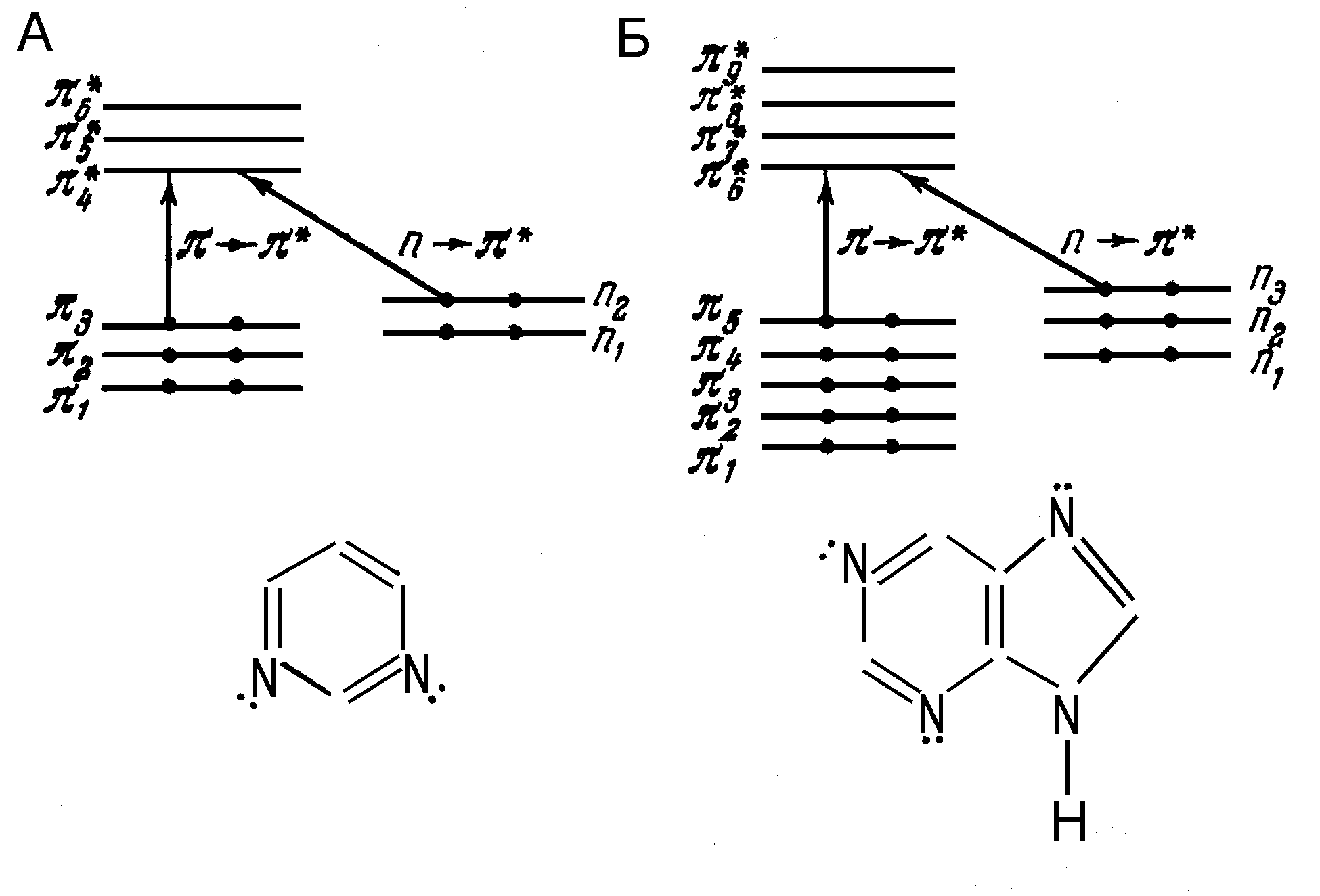
Рис.9. Схема и n уровней пиримидинов (А) и пуринов (Б). Стрелками показаны длинноволновые * и n* переходы. Точки изображают неподеленные пары электронов.
Максимум спектра поглощения всех нуклеотидов лежит в области 260 нм и только у цитидина он несколько смещен к 270 нм. Полоса поглощения света высокомолекулярными ДНК и РНК с максимумом около 260 нм (Рис.3А) представляет собой сумму полос поглощения отдельных нуклеотидов. Эта полоса – визитная карточка нуклеиновых кислот, которые поглощению на длине волны 260 нм можно идентифицировать и измерять их количество.
Оптическая плотность полимерной ДНК примерно на 40 % меньше, чем суммарная оптическая плотности смеси соответствующих нуклеотидов. Этот гипохромный эффект обусловлен пространственной корреляцией отдельных нуклеотидов. Поскольку основной вклад в поглощение на длине волны 260 нм дают переходы, дипольные моменты которых лежат в плоскости молекулы гетероциклов азотистых оснований, то при расположении оснований в параллельных плоскостях они взаимодействует, что снижает вероятности переходов. По гипохромному эффекту нуклеиновых кислот при 260 нм можно оценивать степень их спиральности и ее изменения при действии разных факторов, например, при денатурации. Соответственно, изучая поглощение света при 260 нм, можно следить за ходом денатурации ДНК. Температура денатурации ДНК зависит от средней энергии связей, приходящейся на пару оснований. Так как пары А-Т имеют две, а пары Г-Ц - три водородные связи, то температура денатурации ДНК линейно зависит от процента Г-Ц пар. Поэтому, измеряя спектры поглощения нативной и денатурированной ДНК, можно определить процент Г-Ц пар в ней.
6. Некоторые факторы, влияющие на адсорбционные свойства хромофоров
Хотя основным фактором, определяющим спектральные свойства хромофоров, является его молекулярная структура, окружающая среда: как растворитель, так и микроокружение хромофорной группы в составе биополимера, влияют на спектры поглощения хромофоров. Среди этих факторов важны полярность растворителя, соседних молекул и/или молекулярных группировок в составе белков или мембран, в которые может быть встроен хромофор, их ориентация, рН среды и т.д.
При изменении рН раствора ионное состояние хромофора может изменяться. Например, молекулы, содержащие такие группы как -СООН или -ОН могут диссоциировать. При этом обычно молярный коэффициент поглощения увеличивается и смещается максимум поглощения света. Например, при изменении рН от 6 до 13 у тирозина происходит диссоциация группы –ОН. При этом max увеличивается вдвое, а max смещается от 274 до 295 нм.
Полярность растворителя влияет на полярные группы хромофора, особенно, если в нем есть гетероатомы O, N или S. Обычно при замене полярного растворителя на неполярный max сдвигается вправо, в красную область спектра (батохромный сдвиг).
Параллельное расположение хромофорных групп приводит к гипохромии, как в случае пептидных групп в белках (при 190 нм) или нуклеотидов в двуспиральной ДНК (при 260 нм). Так же возможен спектральный сдвиг. Например, при агрегации молекул хлорофилла появляются, так называемые “нативные формы хлорофилла” со сдвигом max.
Фрайфельдер (1980) сформулировал ряд эмпирических правил, полезных при интерпретации спектров поглощения биологических макромолекул:
1. В неполярном макроокружении молярный коэффициент поглощения аминокислот триптофана, тирозина, фенилаланина или гистидина увеличивается, а max смещается вправо. Поэтому если в водном растворе белка величины и max триптофана или тирозина больше, чем у свободных аминокислот, то эти аминокислоты находятся в неполярном микроокружении внутри белковой глобулы, где они недоступны для воды. Если и max триптофана или тирозина, входящих в состав белка, чувствительны к изменению полярности растворителя, то можно заключить, что аминокислота, для которой наблюдаются изменения этих параметров, располагается на поверхности белка.
У аминокислот, содержащих группы, зарядовое состояние которых может изменяться, таких как –OH группа тирозина или группа –SH цистеина, и max всегда увеличиваются, когда эти группы заряжены. Поэтому если спектр такой аминокислоты не изменяется в условиях, когда, она должна ионизироваться, например, при изменении рН среды, то значит, эта аминокислота спрятана в неполярной области белка. Если же значение рК ионизируемой аминокислоты, определенное по изменению спектра при изменении рН, такое же, как и для свободной аминокислоты в растворе, то она находится на поверхности белка. Но если рК сильно отличается, то эта аминокислота находится в еще более полярном окружении, например, тирозин среди групп -СОО.
3. У пуринов и пиримидинов уменьшается по мере того, как плоскости их молекул становятся параллельными и сближаются друг с другом, т.е. когда возрастает стэкинг-взаимодействие. Поэтому уменьшается в ряду: свободное основание > основание в составе одноцепочечного полинуклеотида без стекинга > основание в составе одноцепочечного полинуклеотида со стекингом > основания в составе двухцепочечного полинуклеотида.
7. Несколько примеров применения абсорбционной спектроскопии
Так как в белках свет поглощают в основном триптофан и тирозин, а содержание их в белках невелико, то изменение спектров поглощения (пертурбация) под влиянием разных факторов (температура, рН, растворитель, химические агенты) может дать ценные сведения о структуре белка, характере связывания коферментов, красителей и других групп, образовании фермент-субстратных и фермент-ингибиторных комплексов. Суть метода состоит в том, что некоторые остатки триптофана и тирозина могут находиться на поверхности белковой глобулы. Их называют доступными, и они обычно подвергаются действию разных внешних факторов. Другие ароматические остатки спрятаны внутри глобулы и не подвергаются влиянию растворителя. Полная их доступность достигается при разрушении конформации белка, например, при денатурации. Отношение числа доступных остатков к общему числу может служить характеристикой конформации белка в данном растворителе. Для оценки числа доступных аминокислотных остатков в раствор белка добавляют “пертурбант” – органическое вещество, с меньшей полярностью, чем вода, например, глюкозу, сахарозу, полиэтиленгликоль, диметилсульфоксид, этанол или глицерин в концентрации около 20 %, которое не слишком изменяет конформацию макромолекулы, но дает реально измеряемый сдвиг полосы поглощения за счет изменения полярности среды. Прецезионные дифференциальные спектрофотометры позволяют зарегистрировать спектральный сдвиг на 0,1-1 нм. В них в один канал помещают кювету с раствором белка без пертурбанта, а в другой – такую же кювету, но с пертурбантом, и регистрируют разностный спектр. Этот метод очень чувствителен, он позволяет зарегистрировать спектральные сдвиги до 10-5 D. Например, для -химотрипсиногена показано наличие 3 доступных и 4-х спрятанных триптофанилов.
Дифференциальная спектрофотометрия также может использоваться для исследования активных центров ферментов. Например, при добавлении различных субстратов к лизоциму наблюдается красный сдвиг полосы поглощения триптофана. Его величина соответствует сдвигу при перемещении одного триптофанового остатка из водной среды в неполярную. Это позволяет предположить присутствие триптофана в центре связывания субстрата. Данные такого несложного спектрофотометрического анализа были подтверждены результатами рентгеноструктурного исследования.
В молекулу белка можно ввести дополнительную хромофорную группу, имеющую спектр, отличный от спектра собственных белковых хромофоров. Изучая изменение ее спектральных свойств, можно судить об изменениях структуры белка. Такие группы называются репортерными. Например, к тирозину в активном центре карбоксипептидазы можно химически присоединить арсаниловую кислоту, спектр поглощения которой очень чувствителен к ионам цинка. По этим данным определили, что центр связывания цинка находится вблизи от активного центра фермента. Интересно, что по данным рентгеноструктурного анализа эти центры находятся на значительном расстоянии. Следовательно, структура этого белка в кристаллах, используемых для рентгеноструктурного анализа, существенно отличается от его структуры в растворе.
ЛИТЕРАТУРА
Миронов А.В.: Основы астрофотометрии. - М.: Физматлит, 2008
Селеменев В.Ф.: Меланоидины. - Воронеж: Воронежский государственный университет, 2004
Э.Н. Аксенова, О.П. Андрианова, А.П. Арзамасцева и др.; Под ред. А.П. Арзамасцева: Руководство к лабораторным занятиям по фармацевтической химии. - М.: Медицина, 2011
И.Я. Гурецкий, В.В. Кузнецов, Л.Б. Кузнецова и др.; Под ред. О.М. Петрухина: Практикум по физико-химическим методам анализа. - М.: Химия, 2007
Кондратьев К.Я.: Аэрокосмические исследования почв и растительности. - Л.: Гидрометеоиздат, 2006
Под ред. И.Н. Глушневой ; И.Б. Волошина, И.Н. Глушнева и др. : Спектрофотометрия ярких звезд. - М.: Наука, 2002
Джадд Д.: Цвет в науке и технике. - М.: МИР, 2008
Берштейн И.Я.: Спектрофотометрический анализ в органической химии. - Л.: Химия, 1975
Раков А.В.: Спектрофотометрия тонкоплёночных полупроводниковых структур. - М.: Советское радио , 1975
Булатов М.И.: Практическое руководство по фотоколориметрическим и спектрофотометрическим методам анализа. - Л.: Химия, 1968
I
1 2 , нм
1
2
3
1 Существующие приборы с кварцевой оптикой не позволяют регистрировать излучение с длиной волны менее 185-190 нм. В этой области также сильно возрастает поглощение света воздухом и водой.
2 CCD (Charge-Coupled Device), по русски – прибор с зарядовой связью (ПЗС).
Поглощение света