Основные теории возникновения и развития опухолей
Контрольная работа
По онкологическим заболеваниям
Тема: Основные теории возникновения и развития опухолей
Содержание
1. Общие сведения о природе опухолей и канцерогенезе
2. Теории онкогенеза
2.1 Мутационная теория рака
2.2. Эпигенетическая теория рака
2.3. Теория химического канцерогенеза
2.4. Хромосомная теория рака
2.5. Вирусная теория рака
2.6. Иммунная теория рака
2.7. Теория раковых стволовых клеток
2.8. Эволюционная теория рака
3. Метастазирование опухолей
Литература
1 Общие сведения о природе опухолей и канцерогенезе
Опухоль возникает только в тканях, способных к пролиферации.
Выделяют два вида опухолей - доброкачественные и злокачественные. Если первые (к числу которых относятся, например, бородавки и жировики), развиваясь, стремятся отделиться от остального организма, то вторые, очень быстро размножаясь, проникают в окружающие ткани, нарушая их естественное развитие, а также образуют новые колонии клеток в отдаленных от исходной опухоли меастах - метастазы.
К доброкачественным опухолям относятся аденомы (лат. adenoma от др.-греч. — железа и –) – продукт доброкачественной неоплазии железистого эпителия. Аденомы встречаются во всех органах, содержащих железистый эпителий и, как правило, гормон-зависимы. В ряде случаев происходит онкогенная трансформация клеток аденомы, что приводит к развитию злокачественной аденокарциномы, как, например в случае колоректального рака, рака простаты и др. (рис. 1).
Рисунок 1 Прогрессирование колоректального рака (КРР). Развитие КРР начинается от предраковых поражений, называемых очагами аберрантных крипт, берущих начало из нормальных клеток слизистой оболочки толстой кишки, далее прогрессирующих до предраковых поражений (аденомы), а затем до инвазивных аденокарцином, которые, наконец, развиваются в метастатические аденокарциномы.
Злокачественные опухоли могут развиваться из эпителиальных и неэпителиальных клеток (клетки кости, крови, мышц). Первые называют раком или карциномой (др.-греч. —«краб», - от — «опухоль»), вторые — саркомой. Некоторые клетки (в частности, кардиомиоциты, клетки волокон хрусталика, рецепторные клетки сетчатки) не способны к пролиферации (делению). Они не замещаются на протяжении всей жизни. Такие клетки исключительно редко подвергаются опухолевой трансформации.
Развитие опухолевого фенотипа представляет собой многостадийный процесс, обусловленный накоплением генетических нарушений. Следствием таких нарушений является постепенное приобретение опухолью все более злокачественного фенотипа. Этот процесс, получивший название опухолевой прогрессии, является важным свойством злокачественных новообразований различного происхождения. Он всегда определяется свойствами исходной ткани, давшей начало опухоли.
Онкотрансформация или злокачественное перерождение тканей основана на дедифференцировке клеток, вызванной нарушением регуляции деления клеток.
Осознание многостадийной природы опухолевого превращения клетки пришло сравнительно недавно, и только в последнее десятилетие стало возможным понять молекулярную природу происходящих при этом событий.
Нормальной клетке необходимо приобрести значительный ряд свойств, для того, чтобы начать формировать выявляемую клинически опухоль. Некоторые из этих свойств, требуют изменений в биохимических процессах на клеточном уровне, другие же требуют нарушения гомеостаза (относительного постоянства внутренней среды организма). Большинство типов опухолей имеет моноклональное происхождение, они развиваются из одиночной клетки. Только индивидуальная клетка сумевшая приобрести все необходимые функции может сформировать центр злокачественного роста. Неспособные к этому клетки погибают либо поглощаются в ходе роста злокачественного клона.
Выделяют четыре стадии канцерогенеза:
1 стадия – стадия инициации.
Первичное воздействие канцерогенных факторов на клетку приводит к возникновению трансформирующего изменения протоонкогенов, т.е. генов, кодирующих белки способные стимулировать образование опухоли, а также выключению генов-супрессоров опухоли (антионкогенов). При этом внутриклеточные сигнальные каскады устроены таким образом, что нарушение лишь одного из звеньев вызывает апоптотическую смерть клетки, прерывая процесс бесконтрольного деления. Для успешного канцерогенеза необходимо изменение многих звеньев, нескольких белков, нескольких генов, максимально имитирующих влияние цитокинов (гормоноподобных соединений, выполняющих регуляторные функции) и способных устранить возможную гибель клетки.
2 стадия – стадия промоции.
В стадии промоции происходит изменения экспрессии ряда сигнальных белков на фоне повторного воздействия на клетку или канцерогенного фактора (того же, что вызвал инициацию, или другого), или фактора, не являющегося канцерогеном, но способного активировать изменённые онкогены, так называемого промотора. Как правило, промоторы вызывают пролиферацию клеток посредством активации пролиферативных сигнальных каскадов, с участием, например, протеинкиназы С. Инициированная клетка приобретает определенные фенотипические свойства только при условии относительно длительного повторного активирующего действия промоторов, что и приводит к необратимой злокачественной трансформации клетки. При этом трансформированные клетки должны приобрести способность обходить иммунный контроль организма «хозяина».
3 стадия – стадия дедифференцировки.
Накопление «несанкционированных» сигнальных белков является хотя и необходимым, но не достаточным условием образования опухоли. Опухолевый рост становится возможным лишь после приобретения способности уклонения трансформированных клеток от дальнейшей дифференцировки. Прекращение дифференцировки возможно при активации генов, некоторых клеточных микроРНК и последующем повреждении участков генома, отвечающих за специализацию клеток. Отсутствие цитокинов, необходимых для перехода созревающих клеток к следующему этапу специализации, также может способствовать прекращению дифференцировки трансформированных клеток (в этом случае присутствие цитокина может вызвать нормализацию и продолжение дифференцировки раковых клеток - процесс, обратный канцерогенезу). Созревание трансформированных клеток приостанавливается, и они, в результате непрерывной пролиферации и подавления апоптоза, накапливаются, формируя опухоль, т.е. клон клеток, обладающих рядом особенностей, не свойственных нормальным клеткам организма. Клетки опухоли с наиболее распространённым набором хромосом образуют стволовую линию.
Геномная неустойчивость, которая вызывает увеличение частоты изменений, необходима для дальней прогрессии опухоли.
4 стадия – стадия прогрессии опухоли.
Биологический смысл этой стадии заключается в окончательном преодолении препятствий на пути роста и распространения опухоли. Сформировавшийся опухолевый клон (стволовая линия) синтезирует собственные цитокины и идёт по пути наращивания темпов деления, уклонения от иммунного надзора организма и обеспечения интенсивного кровоснабжения. Опухолевая прогрессия носит скачкообразный характер и зависит от появления новой стволовой линии опухолевых клеток. В ходе развития опухоли благодаря её генетической нестабильности, происходит частое изменение ее клеточного состава и смена стволовой линии. Подобная стратегия роста носит адаптивный характер, так как выживают только наиболее приспособленные клетки.
Таким образом, в ходе опухолевой прогрессии неопластические клетки приобретают ряд важнейших свойств, которые обеспечивают злокачественный рост опухоли (рис 2).
К этим свойствам относятся:
- иммортализация клеток,
- самодостаточность в пролиферативных сигналах,
- потеря контактного ингибирования,
- дедифференцировка,
- ослабление индукции апоптоза,
- способность к инвазивному росту,
- приобретение локомоторного фенотипа,
- повышение протеолитической активности,
- способность стимулировать ангиогенез,
- генетическая нестабильность.
Что это такое, – описано ниже.
Рисунок 2. Важнейшие свойства, приобретение которых предопределяет способность клетки образовывать злокачественную опухоль (пояснения в тексте).
Иммортализация (от лат. immortalitas — бессмертие) – способность клеток к многократному и даже бесконечному делению, при котором отсутствует репликативное старение. Как известно, существует механизм, ограничивающий число делений большинства типов зрелых клеток человека. Так, в культивируемых in vitro человеческих фибробластах после 60-80 делений (так называемое число Хейфлика) наблюдается необратимая остановка размножения клеток и их постепенная гибель. Между тем, чтобы образовать из одной клетки-родоначальницы сначала опухоль, а затем и метастазы, в условиях жесткого давления со стороны организма, когда многие опухолевые клетки погибают, может потребоваться большее число делений. И, действительно, в опухолевых клетках нарушается работа такого ограничительного механизма контроля репликации. Основными причинами иммортализации раковых клеток являются:
- Искажение клеточного цикла.
- Увеличение продолжительности S-периода клеточного цикла
- Снижение стадии G2 клеточного цикла.
- Клетка вступает в митоз не готовой.
Последствия:
Нарушения при расхождении хромосом.
Высокая потребность в энергии. При этом в злокачественных клетках гликолиз (идущий без кислорода) превалирует над окислительным фосфорилированием.
Самодостаточность в пролиферативных сигналах, т.е. пониженная потребность во внешних сигналах для инициации и поддержания клеточной пролиферации, в первую очередь, белковых факторов роста. При культивировании in vitro большинство типов нормальных клеток размножается лишь при условии, если питательная среда содержит 10-20% сыворотки, т.е. при довольно значительном содержании в ней различных факторов роста. Связывание факторов роста со своими рецепторами инициирует каскады внутриклеточных ферментативных реакций, стимулирующих репликацию ДНК и деление клетки. Оказалось, что многие типы опухолевых клеток способны размножаться в среде с 1% и даже 0,1% сыворотки, т.е. при содержании ростовых факторов в десятки и сотни раз меньшем, чем необходимо для стимуляции размножения нормальных клеток. Такая пониженная потребность в растворимых ростовых факторах достигается изменениями в системах внутриклеточной сигнализации, которые вызывают:
- секрецию необходимых факторов роста самими трансформированными клетками;
- резкое увеличение количества рецепторов для необходимых факторов роста;
- запуск в отсутствие фактора роста каскада событий, аналогичных тому, который в норме инициируется связыванием фактора роста со своим рецептором.
Пониженная потребность неопластических клеток во внешних пролиферативных сигналах также проявляется в так называемой независимости от субстрата, на котором эти клетки растут (anchorage-independence). Большинство типов нормальных клеток способны размножаться лишь при условии их прикрепления к определенному внеклеточному матриксу. Например, фибробласты начинают делиться при взаимодействии с фибронектином. В ином случае пролиферативный стимул, исходящий от растворимых факторов роста, не вызывает полноценного каскада передачи внутриклеточных сигналов, необходимого для стимуляции размножения клеток. Многие типы опухолевых клеток, в отличие от их нормальных предшественников, способны пролиферировать, не прикрепляясь к субстрату, например, в полужидкой среде. Таким образом, неопластические клетки приобретают способность генерировать внутри себя пролиферативные сигналы, в норме исходящие от внешних стимулов.
Потеря контактного ингибирования, т.е. способности к прекращению деления при отсутствии свободного места. Проще говоря, клетки перестают узнавать друг друга. Как известно, в организме определенное число клеток в каждой из тканей поддерживается с помощью разнообразных антипролиферативных сигналов. Такими сигнальными молекулами являются, как цитокины - растворимые белки, секретируемые клетками иммунной системы, так и некоторые белки внеклеточного матрикса и белки на поверхности других клеток. Нормальные клетки, например фибробласты, размножаются до тех пор, пока не возникнет плотный монослой и не установятся межклеточные контакты. В отличие от них, опухолевые клетки обладают пониженной чувствительностью к таким рост-ингибирующим сигналам. При возникновении межклеточных контактов они не останавливают свою пролиферацию, а продолжают делиться, наползать друг на друга и образовывать очаги многослойного роста.
Нарушения дифференцировки, т.е. потери фенотипа, характерного для клеток того или иного типа. Большинство клеток, составляющих разные ткани взрослого организма, дифференцированы. Они приобрели определенный фенотип, свойственный данному органу, например, нейроны в нервной системе, лимфоциты в крови, кардиомиоциты в сердечной мышце. Для многих опухолевых клеток характерны нарушения клеточной дифференцировки. Особенно ярко это проявляется при гемобластозах - новообразованиях из кроветворных тканей, при которых клетки оказываются как бы замороженными на той или иной стадии созревания. Оказалось, что меньшая зрелость лейкозных клеток является не следствием дедифференцировки зрелых клеток, претерпевших неопластическую трансформацию, а обусловлена их происхождением из незрелых клеток, в которых блокированы процессы дальнейшей дифференцировки. Но это свойство не универсально: во многих типах опухолей наблюдается сохранение способности к дифференцировке, причем в отличие от лейкозов, созревание клеток не препятствует приобретению злокачественного фенотипа. Примерами этого могут служить плоскоклеточный рак кожи и высокодифференцированные аденокарциномы толстой кишки, происходящие из незрелых клеток, которые сначала несколько раз делятся, а затем дифференцируются.
В опухолевых клетках могут быть нарушены самые разные компоненты сигнальных путей, ответственных за выполнение дифференцировочных программ.
Ослабление индукции апоптоза. Апоптоз, или запрограммированная смерть клеток, представляет собой активный механизм клеточного самоубийства, поддерживающий в организме определенное число клеток и, кроме того, защищающий его от накопления аномальных клеточных вариантов. Он вызывается как физиологическими сигналами (специфическими «киллерными» цитокинами), так и различными внутриклеточными повреждениями или неблагоприятными условиями, в частности, нарушениями структуры ДНК, нехваткой факторов роста, гипоксией, действием радиации, ультрафиолетового излучения и т.д. Опухолевые клетки способны «ускользать» от апоптоза. Это резко повышает их жизнеспособность, делает их менее чувствительными к факторам противоопухолевого иммунитета и терапевтическим воздействиям.
Способность к инвазивному росту, т.е. к прогрессирующему проникновению в окружающие здоровые ткани (метастазированию). Способность к метастазированию складывается из комплекса приобретаемых клеткой в ходе канцерогенеза ряда признаков, главные из которых: повышение клеточной подвижности и протеолитической активности.
Приобретение способности к миграции связано с изменениями адгезионных взаимодействий клеток друг с другом и с внеклеточным матриксом, и последующими перестройками цитоскелета. Как правило, подвижный или «локомоторный» фенотип неопластических клеток возникает в результате генетических изменений сигнальных процессов, которые у нормальных клеток обеспечивают временное приобретение повышенной миграционной способности. Такие программы, в частности «эпителиально-мезенхимальный переход» клеток эпителия, характерны для эмбрионального развития. Часто в основе возникновения локомоторного фенотипа лежат те же самые изменения, которые вызывают постоянную стимуляцию пролиферации. Дело в том, что верхние и средние этажи сигнальных путей, которые активируются цитокинами, регулируют не только деление, но и движение клеток (являющегося одним из факторов клеточного деления). Например, активация белков семейства Ras и PI3K, находящихся на пересечении сигнальных путей от многих рецепторов, ведет к повышению активности как МАР-киназ и циклин-зависимых киназ – ключевых регуляторов клеточного цикла, так и малых ГТФ-аз семейств Ras и Rho, играющих центральную роль в контроле полимеризации актина, реорганизации цитоскелета и регуляции движения клеток.
Другим важным фактором инвазии опухолевых клеток является их способность продуцировать протеолитические ферменты, которые разрушают окружающий внеклеточный матрикс (например, базальную мембрану эпителиальных органов), создавая тем самым «дороги» для миграции клеток, а также активируют цитокины, стимулирующие миграцию клеток. Протеолитическая активация сигнальных белков, стимулирующих разнообразные факторы транскрипции, приводит к повышению синтеза различных протеаз (например, матричных металлопротеиназ), котрые могут расщеплять Е-кадгерины - белки, осуществляющие межклеточную адгезию, что обусловливает разрушение межклеточных контактов. Однако во многих первичных опухолях во время приобретения инвазивных свойств межклеточная адгезия часто снижена из-за снижения экспрессии E-кадгерина, а затем может снова повышаться в метастазах.
Способность стимулировать ангиогенез - процесс образования новых кровеносных сосудов в органе или ткани. В норме активация ангиогенеза происходит только при росте и развитии организма или при регенерации поврежденных тканей. Интенсивный ангиогенез является важнейшим условием роста опухоли. Это необходимое условие для дальнейшего роста опухолевого узелка, достигшего в диаметре 2-4 мм, т.к. иначе клетки в центре опухоли будут погибать, не получая кислород и питательные вещества. Стимуляция ангиогенеза, т.е. ветвления уже имеющихся в окружающих тканях мелких сосудов и прорастания их в опухоль, вызывается увеличением содержания в микроокружении специфических ангиогенных цитокинов (табл. 1), которые секретируются неопластическими клетками и стимулируют размножение и миграцию эндотелиальных клеток.
Ключевая роль в этом процессе принадлежит эндотелиальному фактору роста сосудов (VEGF), ангиопоэтину-2а, который стимулирует формирование кровеносных сосудов из существовавших ранее, а также таким факторам роста, как основной фактор роста фибробластов (bFGF), плацентарный фактор роста (PLGF), эпидермальный фактор роста тромбоцитов (PD-EGF) и некоторым другим цитокинам. Кроме того, росту новых сосудов способствует уменьшение содержания в микроокружении белков-ингибиторов ангиогенеза, таких как тромбоспондин-1, ангиостатин и эндостатин, а также секреция опухолевыми клетками протеаз, разрушающих внеклеточный матрикс, что необходимо для прорастания новых сосудов.
Для некоторых опухолей, в частности для меланом, характерно образование сети кровоснабжающих трубчатых структур, полностью состоящих из опухолевых клеток – так называемая васкулогенная мимикрия неопластических клеток.
Приобретение способности формировать такие структуры связано с активацией экспрессии в опухолевых клетках ряда белков эндотелиальной дифференцировки, таких как VE-кадгерин (белок клеточной адгезии эндотелия сосудов из семейства кадгеринов, который контролирует и организует межклеточные соединения), VEGFR2 (рецептор 2 сосудистого эндотелиального фактора роста), CD34 (мембранный белок межклеточной адгезии) и др.
Таблица 1 Активаторы и ингибиторы ангиогенеза
|
Активаторы ангиогенеза
|
Ингибиторы ангиогенеза
|
|
Фактор роста эндотелия сосудов (VEGF)
|
Эндостатин
|
|
Плацентарный фактор роста (PlGF)
|
Ангиостатин
|
|
Факторы роста фибробластов (FGF)
|
16 кДа фрагмент пролактина
|
|
Трансформирующие факторы роста a и b
|
Ламинин
|
|
Эпидермальный фактор роста (EGF)
|
Фибронектин
|
|
Инсулиноподобные факторы роста (IGF)
|
Тромбоспондин
|
|
Тромбоцитарный фактор роста эндотелиоцитов (PDECGF)
|
Тромбоцитарный фактор-4 (PF-4)
|
|
Фактор некроза опухолей (TNF, низкие дозы)
|
Фактор некроза опухолей (TNF, высокие дозы)
|
|
Интерлейкины (IL-1, IL-3, IL-6, IL-8)
|
Интерлейкины (IL-12)
|
|
Колониестимулирующие факторы
|
Интерфероны
|
|
Ангиогенин (Ang)
|
Ингибиторы тканевых металлопротеиназ
|
|
Активатор плазминогена урокиназного типа (uPA)
|
Ингибиторы активаторов плазминогена
(PAI-1, PAI-2)
|
Одной из характерных модификаций микроокружения, повышающих инвазивный потенциал клеток, является привлечение в опухоль макрофагов и других клеток, участвующих в воспалительной реакции. При этом в неопластических клетках активируется ряд сигнальных путей, в частности, регулируемых белками Ras (мембраносвязанные белки, участвующие в передаче сигнала от поверхностных рецепторов внутрь клетки), которые обычно регулируют размножение клеток, и NF-B (ядерный фактор транскрипции, контролирующий экспрессию генов иммунного ответа, апоптоза и клеточного цикла), а также путей, стимулирующих продукцию провоспалительных цитокинов (интерлейкинов 1, 6 и 8). Предполагается, что присутствие клеток воспаления ускоряет развитие опухоли за счет секреции ими цитокинов, стимулирующих размножение неопластических клеток, ангиогенез, продукции металлопротеаз, а также повышения содержания активных форм кислорода и азота, индуцирующих мутагенез.
Вероятность возникновения в одной клетке нескольких генетических изменений, придающих совокупность вышеуказанных свойств, резко повышается при нарушениях работы систем, поддерживающих целостность генома. Поэтому мутации, ведущие к генетической нестабильности, являются неотъемлемым этапом опухолевой прогрессии.
Генетическая нестабильность. Генетическая нестабильность неопластических клеток базируется на:
- уменьшении точности воспроизведения генетического аппарата,
- нарушениях механизмов репарации ДНК,
- изменениях регуляции клеточного цикла в поврежденных клетках.
Это, вместе с уходом от апоптоза, позволяющим генетически измененным клеткам выживать, делает популяции опухолевых клеток высоко изменчивой, создает основу для постоянного возникновения и отбора все более и более злокачественных вариантов. Поэтому генетическая нестабильность является двигателем неуклонной опухолевой прогрессии.
2 Теории онкогенеза
Одним из основных вопросов канцерогенеза является вопрос о том, подвергаются ли онкотрансформации одиночные клетки или первоначальный канцерогенный фактор(ы) воздействует(ют) на большое количество сходных клеток?
Моноклональное происхождение новообразований из клона (потомства) одной перерожденной клетки было показано на примере опухолей, происходящих из B-лимфоцитов (B-клеточные лимфомы и плазмоклеточные миеломы), клетки которых синтезируют определенные иммуноглобулины, а также на некоторых других типах опухолей. При этом по мере прогрессирования опухоли из начального клона опухолевых клеток могут развиваться субклоны в результате дополнительных продолжающихся генетических изменений, так называемые «многократные толчки».
Согласно теории «опухолевого поля», сначала поле образуется потенциально неопластических клеток, а затем, в результате размножения одной или большего количества таких клеток может развиться опухоль. При этом от отдельных клональных предшественников может возникнуть несколько обособленных новообразований. Эта теория объясняет происхождение некоторых новообразований в коже, эпителии мочевыводящих путей, печени, молочной железе и кишечнике. Признание факта существования опухолевого поля имеет практическое значение, так как наличие одного новообразования в любом из этих органов должно насторожить клинициста в отношении возможности наличия других подобных новообразований. Например, развитие рака в одной из молочных желез повышает риск возникновения рака в другой приблизительно в 10 раз.
Для объяснения механизмов возникновения как опухолевого моноклона, так и «опухолевого поля» в настоящее время предложен ряд взаимосвязанных концепций:
- мутационная теория рака;
- эпигенетическая теория рака;
- хромосомная теория рака;
- теория раковых стволовых клеток;
- вирусная теория рака;
- иммунная теория рака;
- теория химического канцерогенеза;
- эволюционная теория рака.
2.1 Мутационная теория рака
Согласно мутационной теории, возникновение злокачественных опухолей связано с изменением (мутацией) генома клетки, и в большинстве случаев злокачественное новообразование имеет моноклональное происхождение, т.е. развивается из одной мутировавшей половой или чаще соматической клетки.
Доказательством мутационной природы рака является обнаружение мутаций в протоонкогенах и генах-супрессоров опухолей, вызывающих злокачественную трансформацию клеток. Основные классы генов и их белковых продуктов, которые могут выступить в роли онкогенов или генов-супрессоров опухолей представлены в таблице 2.
Что это такое? Молекулярно-биологическими методами было установлено, что ДНК нормальных эукариотических клеток содержит последовательности, гомологичные вирусным онкогенам, которые получили название протоонкогенов. Протоонкогены являются нормальными клеточными генами. Более того, они участвуют в регуляции важнейших клеточных процессов - клеточного деления, клеточной смерти, репарации ДНК, и их повреждение в результате мутации приводит к неконтролируемому делению клеток и их повышенной устойчивости к апоптозу. Они обладают высокой эволюционной консервативностью, что также подтверждает их важную роль в жизнедеятельности клеток.
Таблица 2. Основные классы онкогенов и генов-супрессоров опухолей
|
Природа гена / белка
|
Ген / белок (примеры)
|
Локализация опухоли (примеры)
|
|
Факторы роста
|
PDGF
|
Глиомы, саркомы
|
|
|
TGF-
|
Многие опухоли
|
|
Рецепторы
|
erb-B
|
Глиобластомы, рак груди
|
|
|
erb-B2
|
Рак груди, яичников, слюнных желез
|
|
Передача сигнала
|
K-ras
|
Рак легких, яичников, кишечника и другие лейкемии
|
|
|
N-ras
|
|
|
Факторы активации
|
c-myc
|
Лейкемии, рак груди, желудка, легких
|
|
Факторы транскрипции
|
N-myc
|
Нейробластомы, глиобластомы
|
|
|
L-myc
|
|
|
Факторы блока
|
TGF-P
|
Рак кишечника
|
|
Передатчики и блокаторы передачи
|
DPC-4
|
Рак поджелудочной железы
|
|
|
NF-1
|
Лейкемии, рак периферической нервной системы
|
|
Контроль клеточного цикла
|
cyclins D, E
|
Рак груди
|
|
|
pl5
|
Разные опухоли
|
|
|
pl6
|
Меланома
|
|
|
pRB
|
Ретинобластома, остеосаркома (наследств.)
|
|
|
p53
|
Многие опухоли (1/2 всех) (наследств.)
|
|
Апоптоз
|
p53
|
Многие опухоли (1/2 всех) (наследств.)
|
|
|
Bcl-2
|
Разные опухоли
|
|
Бессмертие
|
Теломераза
|
Разные опухоли
|
|
Другие гены-супрессоры опухолей
|
APC
|
Рак кишечника (наследств.)
|
|
|
BRCA1, BRCA2
|
Рак груди (наследств.)
|
|
Репарация ДНК
|
Гены репарации
|
Рак кишечника, ксеродерма (наследств.)
|
|
|
ATM
|
Рак груди (наследств.)
|
Можно выделить несколько основных типов мутаций, приводящих к превращению протоонкогена в онкоген.
- Мутация протоонкогена с изменением структуры специфического продукта экспрессии гена приводит к образованию изменённого белка.
Рассмотрим, например, мутации в гене-супрессоре опухолей TP53, кодирующем белок р53. Молекулы белка р53 могут находиться в различных конформационных состояниях (рис.3), выполняя разные физиологические функции.

Рисунок 3. Схематическое изображение различных конформационных состояний р53 (эпитопов), распознаваемых специфическими антителами. Онкогенные мутации вызывают необратимый переход молекулы в денатурированное состояние, при котором открывается ранее недоступный эпитоп и, наоборот, исчезают некоторые ранее доступные эпитопы (по: Б.П.Копнин Опухолевые супрессоры и мутаторные гены (avpivnik.ru/works/new/newinf05_doc).
В обычных условиях белок р53 находится в латентной форме со слабой транскрипционной активностью. При этом он связывает белки, участвующие в репарации ДНК, обладает активностью 3'-5'-экзонуклеазы и стимулирует рекомбинацию и репарацию ДНК. При различных стрессах и внутриклеточных повреждениях могут происходить пост-трансляционные модификации р53, в частности, фосфорилирование и ацетилирование определенных аминокислот, что определяет его переход в так называемую стрессовую конформацию. Такой белок значительно более стабилен, резко увеличивается его количество в клетке, и как фактор транскрипции, он эффективно активирует и/или подавляет экспрессию специфических генов-мишеней, следствием чего являются остановка клеточного цикла и апоптоз. Кроме того, активация белка р53 ведет к изменению экспрессии генов некоторых секретируемых факторов, в результате чего может изменяться размножение и миграция не только поврежденной, но и окружающих клеток. При этом, в стрессовой конформации, способность р53 стимулировать рекомбинацию и/или репарацию ДНК в значительной степени снижается. Основные функции активного белка р53 представлены на рисунке 4.
В белке р53 центральный домен (аминокислоты 120-290) непосредственно узнает и связывает специфические последовательности ДНК регулируемых генов, так называемые р53-реактивные элементы, состоящие из расположенных друг за другом последовательностей с общей структурой типа PuPuC(A/T)(A/T)GPyPyPy (Pu - пурин, Py - пиримидин). Именно в этом ДНК-связывающем домене локализуется большинство точечных мутаций, обнаруживаемых в различных опухолях человека.
Характерные для опухолевых клеток бессмысленные мутации приводят к резкому изменению конформации молекулы белка р53, в результате чего происходит потеря или ослабление способности связывать и активировать гены с р53-реактивными элементами, репрессировать другие специфические гены-мишени, ингибировать репликацию ДНК и стимулировать репарацию ДНК. Причем, так как р53 образует тетрамерные комплексы, мутации в одном аллеле гена ТР53 вызывают инактивацию и продукта второго, неповрежденного аллеля.
Мутации в гене TР53, приводящие к инактивации белка р53, являются наиболее универсальными молекулярными изменениями в различных новообразованиях человека. Более, чем в половине всех опухолей человека (50-60% новообразований более, чем 50 различных типов) обнаруживаются мутации гена TР53. В отличие от других опухолевых супрессоров, для которых характерны мутации, прекращающие синтез белка (делеции, образование стоп-кодонов, сдвиг рамки считывания, нарушения сплайсинга мРНК), подавляющее большинство (более 90%) мутаций TР53 представляет собой бессмысленные мутации, ведущие к замене одной из аминокислот в белковой молекуле на другую.
Рисунок 4. Охранные функции р53. Факторы, вызывающие транскрипционную активацию р53 и биологические эффекты, вызываемые изменениями их экспрессии.
- Другим типом мутаций, приводящих к онкотрансформации клеток, являются точечные мутации регуляторной последовательности протоонкогенов, вызывающие повышение уровня их экспрессии.
Ярким примером таких мутаций является активация протоокогенов семейств ras и raf. Эти гены участвуют в управлении клеточным циклом и являются центральными регуляторами пролиферации и выживания клеток. Точечные мутации этих генов в онкотрансформированных клетках приводят к постоянной стимуляции пролиферации клеток, что способствует росту и инвазии опухоли и развитию метастазов. Мутации одного из генов семейства ras: H-ras, K-ras или N-ras обнаруживаются примерно в 15 % случаев злокачественных новообразований у человека. У 30 % клеток аденокарцином лёгкого и у 80 % клеток опухолей поджелудочной железы обнаруживается мутация в онкогене ras, что ассоциируется с плохим прогнозом протекания заболевания. Мутации генов ras и raf , например, наблюдаются в более 90% клинических случаев меланомы человека. Существуют 3 основные формы мутаций в гене raf: A-raf, B-raf, C-raf. Формирование B-raf мутации играет ключевую роль в патогенезе меланомы. Мутантный белок BRAF постоянно активирует митоген-активируемые протеинкиназы ERK, которые регулируют клеточный цикл. Это стимулирует пролиферацию клеток. Подобные мутации наблюдаются приблизительно в 60-70 % первичных меланом и в 40-70 % случаев метастатических меланом. При этом B-raf мутации вовлечены в инициирование, но не в прогрессию меланом. Мутация V600E, при которой глутамат в положении 600 заменяет валин, найдена в 80-90 % всех B-raf мутаций в меланоме; тогда как мутации в A-raf и C-raf при меланоме наблюдаются редко. Мутации в генах N-ras и B-raf также регулируют экспрессию субъединиц интегринов, что приводит к повышению инвазии клеток меланомы и васкуляризации опухоли, т.е. развитие в ней капиллярной сети.
- Перенос гена в активно транскрибируемую область хромосомы (хромосомные аберрации).
Потеря участка хромосомы, содержащего гены-супрессоры, ведет к развитию таких заболеваний, как ретинобластома, опухоль Вильмса и др.
Функции генов-супрессоров противоположны функциям протоонкогенов. Гены-супрессоры тормозят процессы клеточного деления и выхода из дифференцировки, а также регулируют апоптоз. В отличие от онкогенов, мутантные аллели генов-супрессоров рецессивны. Отсутствие одного из них, при условии, что второй нормален, не приводит к снятию ингибирования образования опухоли, и в ряде случаев инактивация генов-супрессоров ведет к развитию онкологических заболеваний.
Таким образом, система протоонкогенов и генов-супрессоров формирует сложный механизм контроля темпов клеточного деления, роста, дифференцировки и программируемой гибели.
В настоящее время получены многочисленные подтверждения мутационной (генетической) теории рака. Однако, известно, что частота спонтанных мутаций отдельных генов человека в расчете на один ген крайне низка и составляет около 10–5, т.е. одна мутация на 100 тысяч генов. Суммарно частота доминантных мутаций в популяциях человека равна 1%, рецессивных – 0,25% и мутаций хромосом – 0,34%. Доля людей с врожденными дефектами, которые могут проявляться в разных возрастах, составляет около 11%. При этом для возникновения и дальнейшего развития опухоли недостаточно одной мутации, необходимо несколько разных мутаций. В большинстве случаев для полного превращения нормальной клетки в опухолевую в ней должно накопиться порядка 5-10 мутаций. Последние исследования показывают, что прогрессия опухоли определяется не только генетическими, но и эпигенетическими изменениями, которые возникают значительно чаще, чем истинные мутации. Оказалось, что именно ряд эпигенетических изменений во многом способствует дестабилизации генома и большей вероятности возникновения мутаций в генах.
2.2 Эпигенетическая теория рака
Изучение эпигенетических механизмов онкогенеза – активно развивающаяся в последние годы область научных исследований. Эпигенетика (от греч. -над, выше, внешний и генетика) изучает закономерности изменения экспрессии генов и фенотипа клетки, вызванные механизмами, не затрагивающими генетическую информацию, заключающуюся в последовательности нуклеотидов в ДНК.
Основными эпигенетическими процессами являются:
Процесс метилирования ДНК заключается в присоединении метильной группы к цитозину. Обычно это происходит в динуклеотидных группах цитозин-гуанозин: CpG, где p – фосфатная группа, связывающая эти нуклеотиды (рис 5).

Рисунок 5. Выключение» генов осуществляется при помощи метилирования цитозиновых оснований ДНК, прикрепления к ним метильной группы -СН3.
Метилирование цитозина в парах CpG, находящихся в промоторах генов (регуляторных участках), препятствует транскрипции гена, т.е. реализации генетической информации, а деметилирование, наоборот, способствует экспрессии генов. Установлено, что даже незначительные изменения в уровне метилирования ДНК могут существенно влиять на уровень генетической экспрессии. При этом метильная группа выполняет роль «предохранителя». Чем меньше метильных групп в промоторах генов, тем более клетка дифференцирована. Чем выше степень метилирования ДНК, тем ниже степень дифференцировки. Гиперметилирование промоторных областей может подавлять экспрессию генов-супрессоров опухолевого роста, а деметилирование – активировать экспрессию онкогенов. Повышение уровня метилирования генов-супрессоров опухолевого роста в раковых тканях в сопоставлении с нормальными клетками иногда достигает 100%. Доказано, что развитие онкопатологии может быть остановлено при изменении метилирования определенных генетических сайтов в раковых клетках. Определение специфических профилей метилирования в ряде случаев позволяет прогнозировать развитие рака. Глобальное деметилирование ДНК обычно связывают с хромосомной нестабильностью раковых клеток. Однако в них одновременно может наблюдаться гиперметилирование определенных промоторов генов-супрессоров рака.
Гистоны - белки, упаковывающие ДНК в нуклеосомы, из которых формируется ядерный хроматин. Из них гистоны H2A, H2B, H3 и H4 образуют сердцевину нуклеосомы, на которую наматывается ДНК, а гистон H1 связывает нуклеосомы между собой. N-концевые хвостики гистонов могут подвергаться посттрансляционной модификации: ацетилированию, метилированию, фосфорилированию и др. (рис 6). Ацетилирование гистонов приводит к разрыхлению хроматина, и, соответственно, облегчению транскрипции, повышению экспрессии генов и активации синтеза соответствующих белков. Напротив, их деацетилирование связано со снижением транскрипционной активности. Известно, что у раковых клеток снижен уровень ацетилирования гистона Н4 по лизину в положении 16 (K16-H4). Уровень ацетилирования лизина 9 в гистоне Н3 (K9-H3) изменяется при развитии карцином легких, лимфом и сарком мягких тканей мышей, а также при раке легких, простаты и лейкемии у людей. Уровень триметилирования лизина 20 в гистоне H4 (K20-H4) в раковых клетках обычно снижен.
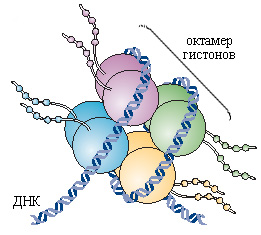

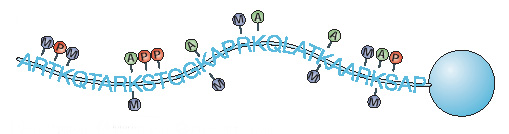
Рисунок 6. Структура нуклеосомы. Гистоны H2A, H2B, H3 и H4, на которые наматывается ДНК, имеют свободные С- и N-терминальные хвосты. Их посттрансляционная ковалентная модификация может осуществляться в результате ацетилирования лизиновых остатков, метилирование лизиновых и аргининовых остатков, убиквитинирование лизиновых аминокислотных остатков, фосфорилирования сериновых и треониновых остатков, а также ADP-рибозилирования остатков глутаминовой кислоты. Это позволяет регулировать транскрипционную активность соответствующих участков ДНК.
- Регуляция генов на уровне РНК (siРНК, микроРНК).
В последнее время большое внимание специалистов привлечено к изучению роли малых интерферирующих РНК (siRNA) в регуляции генетической активности. Интерферирующие РНК могут изменять стабильность и трансляцию мРНК путем моделирования функций полирибосом (несколько рибосом, одновременно транслирующих одну молекулу РНК) и структуры хроматина.
Относительный вклад генетических и эпигенетических факторов в конкретные формы опухолей варьирует в широких пределах.
Недавние исследования привели, однако, к конвергенции этих, казалось бы, непримиримых теорий онкотрансформации клеток. Оказалось, что важную роль в канцерогенезе играют обе составляющие: генетическая и эпигенетическая. Гены-супрессоры и гены репарации ДНК в опухолях могут инактивироваться в результате либо повреждения, либо метилирования промоторов (в последнем случае говорят об «эпимутациях», как об эпигенетическом эквиваленте мутаций, вызывающих такие же функциональные последствия). Становится очевидным, что канцерогенами могут быть не только мутагены, но и другие факторы, воздействующие на клеточный метаболизм.
Давно признано, что одной из основных проблем в терапии рака является неоднородность (гетерогенностью) опухоли, и в частности, наличие нескольких субпопуляций раковых клеток, которые могут обладать свойствами стволовых клеток. Причиной неоднородности клеток опухоли отчасти могут являться вариации эпигеномной структуры хроматина (эпигенома) в клетках-предшественниках опухолей.
Многие вещества имеют свойства эпигенетических канцерогенов: они приводят к увеличению частоты возникновения опухолей, не проявляя при этом мутагенного эффекта (например: арсенит диэтилстилбестрола, гексахлорбензол, соединения никеля). Однако до сих пор не выяснено, как влияют факторы среды на «гистоновый код» и на метилирование ДНК.
В 2008 году Национальный Институт Здоровья США объявил, что в течение следующих 5 лет будет потрачено 190 миллионов долларов на изучение эпигенетики. По мнению некоторых исследователей, эпигенетика может играть большую роль в лечении заболеваний человека, чем генетика.
2.3 Теория химического канцерогенеза
Эта теория рассматривает химические факторы внешней среды как основную причину клеточных мутаций, приводящих к развитию опухоли. Канцерогеном называют фактор, воздействие которого достоверно увеличивает частоту возникновения опухолей (доброкачественных и/или злокачественных) в популяциях человека и животных и/или сокращают период развития этих опухолей.
Канцерогены разделяются на две главных группы: генотоксические канцерогены, которые реагируют непосредственно с ДНК, и эпигенетические, вызывающие изменения ДНК и хроматина без изменения самой последовательности ДНК. Все канцерогены имеют общие свойства:
Их эффекты стойкие, кумулятивные (накапливающиеся) и запаздывающие.
2. Для возникновения опухолей раздельные дозы канцерогена более эффективны, чем одна большая доза.
Внешние канцерогенные факторы можно разделить на три основные группы: физические, химические и биологические. Механизмы действия внешних канцерогенных факторов на клетку пока окончательно не выяснены. А потому отсутствует возможность получения убедительных доказательств их канцерогенного влияния на организм человека, за исключением разве что аналитических эпидемиологических исследований.
В экспериментах на животных выявлено большое количество потенциально канцерогенных химических веществ: ароматические углеводороды (бензпирен, бензантрацен), ароматические амины (анилиновые красители - нафтиламин), некоторые азотистые соединения, ядохимикаты (гербициды, инсектициды), минеральные удобрения, флавоноиды, асбест и т.п. Источником большинства канцерогенов в окружающей среде являются промышленные выбросы. Через загрязненные грунт, воду, воздух, биологические организмы канцерогены попадают на кожу, в легкие, а с пищевыми продуктами - в внутреннюю среду организма. Канцерогенные вещества также образуются при сгорании табака и вдыхаются при курении.
Из физических факторов особого внимания заслуживают различные виды излучений. В результате ядерных испытаний, аварий на атомных электростанциях, атомных кораблях и подводных лодках, расширение сферы деятельности человека, связанной с использованием источников излучения, больших масштабов приобрело распространение радионуклидов. Они могут попадать в организм с питьевой водой, продуктами питания. А поскольку период полураспада основных радиоактивных элементов (кобальт, цезий, стронций) исчисляется десятками лет, то патогенное действие их в организме хроническое, долго действующее. К этому следует добавить, что вследствие уменьшения озонового слоя атмосферы Земли и повышения солнечной активности увеличивается поток и активность ультрафиолетовых лучей.
Все лучевые и ионизирующие воздействия влияют преимущественно на стадию инициации опухоли. При этом их канцерогенное действие может проявиться через несколько лет и даже десятилетий. Другие канцерогенные факторы, например, иммуносупрессоры влияют главным образом на развитие опухоли, и их канцерогенное воздействие может проявиться уже через несколько месяцев. Однако различия между этими видами канцерогенного воздействия трудно уловить вследствие многофакторности таких влияний.
Все известные к настоящему времени канцерогены являются генотоксическими. В результате взаимодействия канцерогенов с ДНК происходит активация протоонкогенов, что является основным механизмом инициации канцерогенеза. На этой стадии канцероген или его активный метаболит взаимодействуют с нуклеиновыми кислотами (РНК, ДНК) и белками клетки. Повреждения клеточных структур, возникающие вследствие такого взаимодействия, обычно носят генетический характер (изменения в последовательности ДНК или числа хромосом, генные/эпигенетические мутации, хромосомные аберрации и т.д.). После того, как произошли изменения в генетическом аппарате клетки, наличие канцерогенного воздействия для дальнейшего развития неопластического процесса уже не является обязательным.
Однако следует подчеркнуть, что клетки обладают сложной системой репарации повреждений ДНК, вызываемых разнообразными агентами химической и физической природы. Эффективное функционирование этой системы может обеспечить сохранение нормального генотипа клетки даже в условиях, когда клетка подвергается воздействию канцерогенных факторов, поэтому влияние канцерогена наиболее значимо в случае повреждения клеточных систем репарации ДНК.
2.4 Хромосомная теория рака
В 1999 г. Питер Дюсберг из Калифорнийского университета в Беркли создал теорию, согласно которой рак является следствием исключительно анеуплоидии, а мутации в специфических генах не играют значимой роли в канцерогенезе.
Термин «анеуплоидия» использовался для описания изменений, вследствие которых клетки содержат число хромосом, не кратное основному набору, но в последнее время его стали применять в более широком смысле. Теперь под анеуплоидией понимают также укорочение и удлинение хромосом, перемещение их крупных участков (транслокации). Большинство анеуплоидных клеток сразу же погибают, но у немногих выживших тысячи генов оказываются не такими, как у нормальных клеток. Слаженная совокупность ферментов, обеспечивающих синтез ДНК и её целостность, распадается, в двойной спирали появляются разрывы, ещё больше дестабилизирующие геном. Чем выше степень анеуплоидии, тем менее стабильна клетка и тем больше вероятность того, что, в конце концов, появится клетка, способная расти где угодно. В отличие от трех предыдущих теорий, гипотеза изначальной анеуплоидии полагает, что зарождение и рост опухоли в большей степени связаны с ошибками в распределении хромосом, чем с возникновением в них мутаций.
Впервые характерные для опухолей изменения кариотипа (набора хромосом определенной структуры, специфической для данного организма) были обнаружены в клетках хронического миелоидного лейкоза (ХМЛ) - рака крови, обусловленного неконтролируемым делением клеток в костном мозге. В этих клетках в результате переноса длинного плеча хромосомы 22 на длинное плечо хромосомы 9 появляется так называемая филадельфийская (Ph') хромосома. Рh'-хромосома, как и многие другие хромосомные маркеры, не связанные с мутацией в половых клетках, является приобретенным, а не наследуемым признаком.
В последние полтора десятилетия благодаря новым методам приготовления и дифференциальной окраски хромосомных препаратов, позволяющих идентифицировать каждую хромосому в отдельности, специфические изменения кариотипа выявлены в клетках некоторых опухолей человека, главным образом, гемобластозов. При остром миелобластном лейкозе (ОМЛ) происходит транслокация (перемещение) части хромосомы 8 на хромосому 21, или транслокация с 6-й на 9-ю хромосому. Специфические хромосомные изменения обнаружены при острых промиелоцитарном, монобластном и лимфобластном лейкозах, лимфоме Беркитта и т. д. Некоторые специфические нарушения хромосом в костномозговых клетках, отсутствующие в период установления диагноза, появляются в поздние стадии заболевания, т е в ходе прогрессии опухоли, например транслокации между 6-й н 9-й хромосомами при ХМЛ. Однако, для ряда опухолей анеуплоидия не характерна.
2.5 Вирусная теория рака
Обнаружение вирусов в целом ряде злокачественных опухолей, таких как, саркома Капоши, рак шейки матки, рак печени и др. позволило Л.А. Зильберу в 1945 году сформулировать теорию вирусной природы рака. Основным постулатом этой теории является утверждение о том, что геном клетки может нарушаться вследствие активации ингегрированной в него ДНК вируса.
Как известно, вирус представляет собой генетический материал (ДНК или РНК), запакованный в белковую оболочку. Встраиваясь в геном нормальной клетки, он заменяет ДНК (или РНК) клетки, что вызывает генерацию самой клеткой новых копий вируса.
Согласно вирусной теории рака, геном опухолеродного вируса интегрируется в геном нормальной клетки, что вызывает её бесконтрольное деление. Геном вируса, встроенного в ДНК клеток хозяина был назван провирусом. В 70-е годы 20-го столетия в некоторых РНК-содержащих вирусах были обнаружены гены, необходимые для превращения нормальной клетки в опухолевую. Эти гены были названы онкогенами или трансформирующими генами вирусов - v-onc. В дальнейшем копии или аналоги вирусных онкогенов были найдены в геномах нормальных клеток человека и животных, а также была экспериментально доказана способность онкогенов клеток теплокровных встраиваться в геном вируса. В настоящее время большинство онкогенов идентифицировано, установлена их химическая структура, локализация в хромосомах, а также выявлены белки – продукты активности этих генов (табл. 3).
Обнаружено несколько типов вирусов вызывающих возникновение злокачественных опухолей у человека. Среди них вирус папиломы человека (провоцирует развитие рака шейки матки), вирус гепатита В (приводит к гепатоцеллюлярному раку печени), вирус иммунодефицита человека (причина развития саркомы Капоши), вирус Т-клеточного лейкоза человека - ATLV (adult T-cell leukemia virus), вирус Эпштейна-Барра из группы вирусов герпеса, являющийся весьма вероятным этиологическим фактором лимфомы Беркитта и некоторые другие.
Таблица 3. Наиболее значимые онкогены и продукты их активности
|
Протоонкогены
|
Онкогены
|
Функция онкобелка
|
|
Ген PDGF – фактора роста тромбоцитов
|
sis – онкоген вируса саркомы обезьян
|
Ростовой фактор, аналог фактора роста тромбоцитов
|
|
Ген EGF-R – рецептора эпидермального фактора роста
|
erbB – онкоген вируса эритробластоза птиц
|
«Обезглавленный» рецептор фактора роста, непрерывно посылающий сигналы к пролиферации
|
|
C-ras – ген белка, входящего в систему передачи сигнала в клетку
|
ras – онкоген вируса саркомы и многих опухолей животных и человека
|
Цитоплазматический активированный передатчик сигналов в клетку, ведущий к ее пролиферации
|
|
c-src – ген тирозинкиназы, критического звена в системе передачи сигнала в клетку
|
src – онкоген вируса саркомы птиц и млекопитающих
|
Активированный передатчик сигналов в клетку, ведущий к ее пролиферации
|
|
c-myc – ген ядерного транскрипционного фактора
|
myc – онкоген вируса лейкоза птиц и многих опухолей человека и животных
|
Ядерный фактор, активность которого ведет к непрерывному делению клеток
|
Первый онкоген был открыт в 1978 г., его назвали src от слова «саркома». Этот проонкоген кодирует фермент протеинкиназу Src, повышение активности которой превращает нормальные клетки в раковые. Однако при изучении гена src было обнаружено, что этот ген не играет никакой роли в жизни тех вирусов, которые его содержат, но тогда откуда в вирусах взялся ненужный им ген? Оказалось, что ген src не заносится в геном клеток вирусами, а всегда присутствует в клетках человека. Однако в норме этот ген находиться в «молчащем» состоянии, т.е. на нем не синтезируется мРНК и не производится белок. Но если этот ген внесен вирусом, то он начинает работать. Было показано, что включение гена src в геном вируса меняет регуляцию работы этого гена. В геноме вируса перед геном src оказывается очень активный вирусный промотор (последовательность нуклеотидов в ДНК, узнаваемая РНК-полимеразой, начало транскрипции), наличие которого и делает ген src онкогеном. После встраивания его в таком виде в геном нормальной клетки онкоген начинает активно работать, что приводит к синтезу фермента (рис. 7). Необходимые промоторные участки содержатся в больших терминальных повторах ДНК-копий РНК-содержащих вирусов. В роли активатора структурных генов могут выступать мобильные генетические элементы, способные перемещаться по геному и встраиваться в различные его участки. Они называются транспозирующими элементами генома или энхансерами (enchancer – усилитель). Энхансеры активируют транскрипцию структурного гена, зачастую находясь на расстоянии многих тысяч пар нуклеотидов от него, а иногда они могут быть встроены в хромосому после гена. Подобный образом, например, активируется и затем транскрибируется ген тус.
Помимо src в последние годы было обнаружено ещё несколько десятков подобных онкогенов, которые присутствуют в геноме нормальных клеток позвоночных и играют важную роль во время эмбрионального развития, когда необходимо частое деление клеток. По мере развития организма работа этих генов блокируется регуляторными системами клеток. Но когда эти гены начинают активно работать в клетках взрослого организма, происходит значительный рост пролиферации этих клеток и как следствие развитие опухоли.
Рисунок 7. Схема процесса активации протоонкогена в результате вставки промотора.
Прямая роль вирусов в возникновении злокачественных и доброкачественных опухолей человека доказана пока лишь в единичных случаях и концепция о едином механизме канцерогенеза, связанным с провирусами, не нашла своего подтверждения. В настоящее время вирусная теория канцерогенеза рассматривается как частный случай, а общим механизмом в возникновении опухолей считается преобразование собственных клеточных генов (протоонкогенов) в онкогены путем их активации.
Следует отметить, что даже в случаях доказанной вирусной природы опухоли нет оснований подозревать заразность заболевания. Вирусы, которые считаются причиной возникновения некоторых форм рака животных и человека не передаются контактным путем.
2.6 Иммунная теория рака
Согласно иммунной теории, первопричиной рака является не столько возникновение мутантных клеток, сколько нарушение защитных механизмов организма по их обнаружению и разрушению. Известно, что, несмотря на значительное количество мутантных клеток, которые постоянно образуются в организме здорового человека, опухоль развивается далеко не всегда. Кроме того, большая частота возникновения опухолей наблюдается у пациентов, получающих препараты, подавляющие иммунитет, или в случае иммунодефицитов, вызванных другими факторами, например действием канцерогенов или вследствие наследственных нарушений отдельных звеньев иммунной системы. На основании этой теории можно объяснить, почему с возрастом риск развития рака прогрессивно повышается. На фоне возрастного увеличения частоты мутаций, по причине возникающих дефектов в системе репарации ДНК, наблюдается снижение иммунной реактивности.
Однако ряд фактов противоречит данной теории. Например, у мышей с генетически обусловленной недостаточностью Т-клеточного иммунитета частота новообразований не повышается. Иммунодефициты разной этиологии у людей приводят к развитию главным образом лимфом, а не полного спектра злокачественных новообразований, удаление тимуса не повышает частоты возникновения рака. Кроме того, многие опухоли синтезируют опухолевые антигены и вызывают развитие полноценного цитотоксического иммунного ответа организма на них, однако этот ответ часто оказывается неэффективным. У большинства больных раком регистрируются как клеточные, так и гуморальные иммунные реакции против опухолевых антигенов, но трансформированным клеткам удается обходить иммунную защиту организма. Возможными механизмами иммунотолерантности опухолей могут быть:
- Нечувствительность к действию лимфоцитов. Введение высокой концентрации опухолевых антигенов или клеток опухоли до развития полноценного иммунного ответа вызывает развитие специфической ареактивности. Кроме того, в органах с естественно сниженным иммунным надзором, например, в головном мозге, опухоли вообще не подвергаются действию иммунитета.
- Гетерогенность клеток опухоли. Любая опухоль представляет собой гетерогенную популяцию клеток. Если в результате активации иммунной системы организма, часть опухолевых клеток погибает, другие, утратившие собственные, присущие данной опухоли поверхностные антигены или обладающие меньшей чувствительностью к действию лимфоцитов, продолжают активно размножаться, обеспечивая дальнейший рост опухоли. Кроме того, уцелевшие раковые клетки становятся устойчивыми к определенным антителам. Организм продуцирует новые антитела и лимфоциты, но вновь части клеток опухоли удается уйти от разрушения и обеспечить дальнейший её рост, причем приобретая новые свойства. Подобные закономерности наблюдаются и в случае борьбы с инфекциями.
- Маскировка опухолевых антигенов. В ходе иммунных реакций некоторые антигены опухоли меняют свою структуру, тем самым снижая специфичность иммунного ответа. Кроме того, некоторые антигены опухоли покрыты нейтральным слоем полисахаридов, что мешает лимфоцитам распознавать их.
- Иммуносупрессивное действие опухолей. В некоторых случаях опухолевые клетки сами выделяют факторы, угнетающие активность защитных механизмов организма хозяина. Показано, что при лимфогранулематозе клетки опухоли в лимфоузле сами выделяют или стимулируют секрецию макрофагами, Т-клетками факторов, подавляющих клеточные иммунные реакции.
Ряд исследований свидетельствует о том, что во многом именно противоопухолевый иммунитет обеспечивает невозможность контактной передачи рака, даже в тех случаях, когда этот рак имеет вирусную природу. При искусственном введении взрослому животному онкогенного вируса опухоль не только не возникает, но напротив организм приобретает устойчивость к любой опухоли, вызванной этим вирусом. Однако, если этот вирус вводится новорожденному организму с несформированной иммунной системой, может происходить злокачественная трансформация нормальных клеток.
Таким образом, противоопухолевый иммунитет с одной стороны борется с клетками опухоли, а с другой активно способствует возникновению иммунотолерантных раковых клеток. Кроме того, в процессе роста наблюдается образование не только «хороших» антител, разрушающих опухоль, но и так называемых «блокирующих» антител, которые нейтрализуют действие хороших, а также образование антител, наоборот, стимулирующих рост опухоли. Поэтому основная задача противораковой иммунной терапии состоит не столько в подстегивании иммунной системы организма, сколько в смещении баланса сил в сторону эффективной борьбы с клетками опухоли.
2.7 Теория раковых стволовых клеток
В настоящее время большинством ученых занятых проблемой рака высказывается мнение, что теория раковых стволовых клеток (РСК) является лучшим кандидатом для объяснения биологии рака. Как известно, рак - это гетерогенная группа заболеваний с различными биологическими свойствами, которые обусловлены рядом клональных генетических изменений, в основном, в онкогенах и генах-супрессорах опухоли. Однако последние данные свидетельствуют о том, что рак имеет принципиально общую основу, которая базируется на поликлональных генетических/эпигенетических нарушениях стволовых/прогениторных клеток при посредничестве «генов предшественников опухоли» («tumour-progenitor genes»).
Нормальные стволовые клетки можно обнаружить в большинстве тканей взрослого организма. Они обеспечивают рост, обновление и репаративную регенерацию органов и тканей, что особенно важно для постоянно обновляющихся и регенерирующих тканей, таких как кровь и эпителий. Нормальные стволовые клетки относительно не многочисленны и, как правило, находятся в состоянии покоя или медленно делятся. Их многочисленные дочерние клетки напротив активно делятся и дифференцируются, формируя необходимые компоненты тканей.
Интересно, что нормальные стволовые клетки более устойчивы к радиации или химиотерапии, чем их дифференцированное потомство, как полагают, из-за их низкой скорости деления. РСК обладают рядом характерных отличий от нормальных стволовых клеток.
Во-первых, для нормальных стволовых клеток характерно асимметричное деление, в результате которого образуется одна дочерняя стволовая клетка и одна клетка, которой предстоит прекращение деления и терминальная дифференцировка. Если нормальная стволовая клетка будет быстрее делиться, это лишь увеличит скорость обновления клеток, т.к. дифференцированные клетки будут быстрее образовываться и быстрее отмирать, и обе популяции клеток сохранятся в правильном соотношении. В случае РСК в результате трансформации клеток происходит нарушение этого баланса: либо стволовыми клетками остаются более половины дочерних клеток, либо процесс дифференцировки изменен так, что дочерние дифференцирующие клетки сохраняют способность к неограниченному делению (рис. 8).
По определению РСК являются субпопуляцией раковых клеток со способностью, подобной нормальным стволовым клеткам, давать все типы раковых клеток, обнаруживаемые в опухоли. Причем в этой популяции могут быть клетки с довольно дифференцированными признаками. Точное происхождение РСК на сегодняшний день неизвестно. РСК не обязательно происходят из нормальных стволовых клеток и неясно обладают ли они вообще способностью дифференцироваться или другими характеристиками нормальных стволовых клеток. Т.о., название «раковая стволовая клетка» (cancer stem cell) является скорее отражением фенотипа, подобного стволовым клеткам, чем собственно стволовости (stemness, совокупность всех свойств, присущих стволовым клеткам).
Рисунок 8. Стратегия раковой стволовой клетки и её роль в клеточной дифференцировке. Два типа нарушений, которые могут приводить к характерной для рака неудержимой пролиферации. Следует отметить, что повышенная скорость деления раковой стволовой клетки сама по себе не приводит к такому эффекту.
Впервые предположение о существовании клеток такого типа было выдвинуто в 90-х годах в ходе опытов над мышами с лейкемией. Исследователи установили, что популяция очищенных клеток острой миэлоидной лейкемии, экспрессирующих определенные маркеры клеточной поверхности, может эффективно формировать опухоли, будучи инъецированной мышам, тогда как другие клетки из того же самого рака не обладали такой способностью. Затем, используя этот подход, были идентифицированы РСК в опухолях груди, головного мозга, легких, кожи, печени, поджелудочной железы и др.
Однако, долгое время не удавалось доказать существование РСК в естественно развивающихся опухолях. В 2012 г. сразу три группы ученых независимо друг от друга представили убедительные доказательства наличия РСК в опухолях, которые были опубликованы в ведущих научных журналах Nature и Science. Для обнаружения РСК все три группы использовали метод генетического маркирования. Объектом исследования были разные типы раковых опухолей. Ученые из Техаса Chen J. и соавт. (статья в журнале Nuture: «A restricted cell population propagates glioblastoma growth after chemotherapy») проводили исследования на клетках глиобластомы. Им удалось обнаружить в популяции клеток глиобластомы отдельные клетки, меченные трансгенным (трансген – это чужеродный фрагмент ДНК, переносимый при помощи генно-инженерных манипуляций в геном определенного организма) маркером nestin-TK-IRES-GFP, который помечает только покоящиеся нервные стволовые клетки мозга. Другие клетки опухоли данный маркер не содержали. Дальнейшие опыты показали, что при проведении химиотерапии цитостатиком темозоломидом гибнут все клетки, кроме «меченых». Именно эти клетки дают начало новой опухоли. При подавлении процесса активации этих делящихся клеток с помощью ингибитора ДНК-полимераз ганцикловиром удавалось достичь распада опухоли на фрагменты, которые не могли развиться в новую опухоль.
Научная группа из Бельгии Driessens G. и соавт. (статья в журнале Nuture: «Defining the mode of tumor growth by clonal analysis») идентифицировала с помощью клонального анализа в популяции клеток плоскоклеточного рака кожи клетки, способные к неограниченному делению, т.е. - РСК. Для этого была использована стратегия генетической маркировки отдельных клеток опухоли, находящихся на разных стадиях развития рака. Было обнаружено, что большинство меченых клеток рака кожи имеют ограниченный пролиферативный потенциал и в дальнейшем дифференцируются в клетки инвазивного плоскоклеточного рака, в то время как отдельный клон клеток сохраняют способность к делению длительное время и практически не дифференцируются, причем оказалось, что, чем более агрессивна опухоль, тем больше таких клеток образуется.
Ученые из Голландии Schepers A.G. и соавт. (статья в журнале Science: «Lineage tracing reveals Lgr5+ stem cell activity in mouse in intestinal adenomas») работали с клетками, формирующими аденомы – предшественники рака кишечника (колоректальный рак). Они вывели трансгенных мышей, несущих специфический генетический маркер Cre (cAMA чувствительный элемент)-reporter R26R-Confetti: при введении соединения, повышающего внутриклеточный уровень цАМФ клетки кишечника начинали производить молекулы четырех цветов в зависимости от того, какие клетки являлись родоначальниками (рис. 9). Были получены опухоли из клеток разного типа, но окрашенных в один цвет, что доказывает их общее происхождение из одной РСК.
Пока неизвестно, могут ли эти данные быть применимы ко всем видам опухолей, но несомненно одно, проведенные исследования, доказывающие возможность развития опухоли из одной трансформированной стволовой клетки, в корне изменят оценку эффективности того или иного вида противоопухолевой терапии и приведут к дальнейшему развитию направленной (таргетной) терапии рака.
Рисунок 9. После рекомбинации в ходе мейоза каждая клетка начинает непрерывно производить один флуоресцентный белок - GFP (зеленый), YFP (желтый), RFP (красный) или CFP (синий). Возможные дочерние клетки будут производить тот же флуоресцентный белок, создавая клоны клеток с тем же цветом.
В настоящее известно, что как взрослые стволовые клетки, так и их производные клетки-предшественники (дочерние клетки) или даже более дифференцированные клетки могут стать РСК и дать начало опухоли (рис. 10). Взрослые стволовые клетки, присутствующие практически во всех тканях и являющиеся долгоживущими скорее, чем др. клетки способны приобретать множественные мутации, необходимые для онкотрансформации. Было показано, что большинство РСК экспрессируют (синтезируют белки- маркеры), ассоциированные со взрослыми стволовыми клетками, такие как CD133 (проминин-1, клеточная функция до конца неизвестна), CD44 (гликопротеид, который является рецептором для гиалуронана, главного компонента внеклеточного матрикса) и ALDH1 (фермент альдегиддегидрогеназа). Как нормальные стволовые клетки, так и РСК, по-видимому, обладают общими эпигенетическими профилями, профилями генной экспрессии и активированными сигнальными путями, такими как Notch, Hedgehog и Wnt.
Рисунок 10. Взрослые стволовые клетки, клетки-предшественники (дочерние клетки) или дифференцированные клетки могут приобрести генетические и эпигенетические изменения необходимые для раковой стволовой клетки.
Дифференцированные клетки, приобретающие вследствие мутаций свойства клеток предшественников или стволовых клеток, могут стать РСК. Возможно, что взрослые стволовые клетки могут приобретать первичные генетические и эпигенетические изменения и что дополнительные мутации могут накапливаться в клетках-предшествинниках или более дифференцированных дочерних клетках. Источник РСК, комбинация приобретаемых генетических и эпигенетических изменений и влияние микросреды, все это способствует возникновению РСК и скорее всего, предопределяет, какие маркеры РСК будут экспрессировать и какого типа опухоль возникнет из них. Неясно, сохраняют ли РСК все свои характеристики по мере развития опухоли. РСК, выделенные из опухолей человека, могут быть не идентичными тем, которые присутствовали при более раннем развитии опухоли. Т.о., точные молекулярные профили и др. характеристики РСК, участвующие в онкогенезе, неясны. Поскольку маркерные белки появляются на клеточной поверхности нормальных клеток, то таковые должны наблюдаться на поверхности РСК из многих типов тканей, такие как, например, CD133 и CD44. Скорее всего, именно они являются настоящими маркерами РСК, участвующих в онкогенезе, что объясняет их воспроизводимость и их присутствие на поверхности РСК. Однако эти маркеры могут также отражать способность определенных клеток переживать процедуру очистки или инициировать опухолевый рост у мышей. Большинство исследователей отмечает значительные ограничения подхода трансплантации раковых клеток мышам, используемого для идентификации РСК, т.к. раковые клетки человека трансплантируются нормальным мышам без потенциально важных акцессорных клеток (в основном макрофаги и фибробласты) и при нарушении и несовместимости иммунной системы.
Раковые стволовые клетки, как полагают, участвуют в опухолевом росте, но участие определенных типов РСК в этом процессе и значение разных процентов раковых стволовых клеток, обнаруживаемых в опухолях, неясно (рис. 11).
Рисунок 11 В ходе роста опухоли, любая раковая клетка может приобрести новые генетические или эпигенетические мутации или изменяться благодаря влиянию микросреды, в результате эти изменения приведет к возникновению нового типа раковых стволовых клеток. Разные типы раковых стволовых клеток в опухоли образуют новые популяции (клоны) клеток разных размеров, которые содержат как раковые стволовые клетки, так и клетки разной степени дифференциации. Общий процент клеточных подтипов в опухоли, в которые входят и раковые стволовые клетки разного фенотипа может определять исход заболевания.
Наиболее вероятно, что в одной опухоли может присутствовать несколько популяций РСК. Т.к. раковые клетки постоянно эволюционируют посредством возникновения новых генетических и эпигенетических изменений и благодаря влиянию на них микроусловий, то потенциально любая из них может стать РСК. Также сами РСК могут приобретать новые свойства, как это показано для РСК при лейкемии, у которых наблюдаются перестройки генов иммуноглобулинов. Паттерны мутаций, которые можно обнаружить в разных регионах опухоли, свидетельствуют о том, что имеются популяции клональных раковых клеток, и некоторые из них могут быть раковыми стволовыми клетками. Необходимо использование большого количества различных маркеров для идентификации РСК даже внутри одиночной опухоли.
Процент РСК от общего количества клеток опухоли может варьировать от 0.03% до почти 100%, что связано со свойствами РСК, инициировавших развитие опухоли и микроусловиями, которые детерминируют возможность возникновения новых РСК. Присутствие большого количества РСК в опухоли указывает на высокий пролиферативный потенциал опухоли, её генетическую нестабильность и отсутствие в ней дифференцировки, что обуславливает эффективность лечения и исход заболевания.
Предполагается, что большая гетерогенность популяции клеток опухолей, является одной из причин их высокого метастатического потенциала, большой вероятности ремиссий и низкой эффективности лечения.
2.8 Эволюционная теория рака
Согласно эволюционной теории рака, формирование опухоли представляет собой эволюционный процесс путем природной селекции клонов клеток, приобретающих наследуемые характеристики, дающие им преимущества. Впервые эта теория была предложена Новелом в 1976 г. Он предположил, что природная селекция опухолей основана на клональной селекции отдельных клеточных клонов, что приводит к постоянному изменению характеристик клеток. При этом главным показателем способности опухоли эволюционировать является степень наследственной вариации клеток внутри опухоли. Основой подобной вариации клеток являются соматические мутации клетки-прародителя, дающей начало опухоли, а также огромное число индивидуальных, характерных для отдельных клеток генетических и эпигенетических изменений, которые приобретаются отдельными клетками опухоли во время деления и формирования дочерних клеток. Т.е. предполагается, что опухоль имеет моноклональное происхождение из раковых стволовых прогениторных клеток. Однако с ростом размера опухоли наблюдается расхождение популяции опухолевых клеток на субклоны, что является основой гетерогенности опухолей. При этом степень гетерогенности опухоли на генном уровне служит основой эволюции опухоли как системы, поскольку она определяет пластичность опухоли и больший адаптационный потенциал, что является механизмом, обеспечивающим резистентность опухоли к терапии. Причем субклоны клеток с высокой способностью к формированию злокачественных новообразований характеризуются высокой частотой неклональных изменений по сравнению с клетками опухолей с низкой онкогенностью.
3 Метастазирование опухолей
Метастаз – это отдалённый вторичный опухолевый очаг, образовавшийся в результате перемещения клеток из первичной опухоли через ткани организма. Новые очаги рака, метастазы, вызывают дисфункцию органов и смерть.
Метастазирование складывается из следующих этапов:
- Открепление раковой клетки от опухоли вследствие ослабленной адгезии;
- проникновение опухолевых клеток через сосудистую стенку в просвет кровеносного или лимфатического сосуда;
2) перенос опухолевых клеток током крови или лимфы;
3) прикрепление опухолевых клеток к стенке сосуда на новом месте;
4) выход опухолевых клеток через сосудистую стенку в окружающую ткань;
5) размножение клеток и формирование вторичного опухолевого очага;
6) рост метастаза.
Как происходит подготовка к метастазированию? Есть две основные модели:
1) Согласно генетической модели, мутации в некоторых клетках опухоли повышает их способность к инвазии и миграции этих клеток к новому месту, где они делятся, образуя метастазы. Поскольку генетические повреждения необратимы, то метастазы генетически отличаются от первичной опухоли. Так, в меланоме выявлены мутации в генах, связанных с метастатическим процессом. Было установлено, что активация гена NEDD9 в меланоме связана с метастазированием. Другим заметным событием является повышение числа копий хромосомы 6р. Амплификация этой хромосомы наблюдается более, чем в >35% метастатических образцов и клеточных линий меланомы, что связано с плохим прогнозом. Но она редко обнаруживается в первичной меланоме, Если предположить, что возникновение метастазов действительно происходит за счет приобретения необратимых генетических изменений, то удивительно, почему повышенная экспрессия именно этих генов до сих пор не обнаружена во всех метастазах данной опухоли.
2) Альтернативная модель тесно связана с теорией стволовых клеток и предполагает, что метастазы возникают в результате влияния генетических/эпигенетических мутаций и микроокружения, что приводит к изменению фенотипа клеток опухоли и появлению отдельных пролиферирующих клеток – метастатических раковых стволовых клеток.
Как могут появляться метастатические стволовые клетки?
Возможно, что исходные РСК, давшие начало опухоли приобретают дополнительные свойства, необходимые для метастазирования, за счет генетических/эпигенетических изменений и влияния микросреды опухоли. Т.е. развитие первичной и метастатической опухолей идет параллельно, а не последовательно (рис. 12).
Рисунок 12. Начало метастазам могут давать раковые стволовые клетки, инициировавшие развитие первичной опухоли или новая популяция раковых стволовых клеток с дополнительными клетками опухоли или без них. Новые раковые стволовые клетки могут образовываться благодаря генетическим/эпигенетическим изменениям, влиянию микроокружения и в ходе эпителиально-мезенхимального перехода.
В другом случае, наблюдается возникновение нового типа РСК, обладающими метастатическими свойствами. Благодаря дополнительным генетическим или эпигенетическим альтерациям эти клетки приобретают определенные преимущества над исходными РСК, т.е. приобретают свойства, обуславливающие их высокую инвазивную способность, чем у исходных РСК и легко метастазируют (Рис. 12). Формирование подобных метастатических стволовых клеток можно наблюдать в случае так называемого эпительально-мезенхимального перехода (epithelial-mesenchymal transition (EMT)) на инвазивном крае опухолей. Более того, недавно было установлено, что EMT наделяет клетки свойствами раковых стволовых клеток и что предполагаемые стволовые клетки рака груди экспрессируют маркеры EMT, что свидетельствует о связи между EMT и раковыми стволовыми клетками. Кроме того, определенные субпопуляции раковых стволовых клеток обнаружены на инвазивном крае панкреатических карцином, что является важным признаком возможности метастазирования опухоли.
Наконец, раковые стволовые клетки могут метастазировать вместе с др. типами раковых клеток.
На сегодняшний день качественные особенности метастатических раковых стволовых клеток, а также механизмы, с помощью которых они заселяют метастазы, во многом неясны.
Было показано, что метастазы содержат небольшой процент раковых стволовых клеток и более высокий процент более дифференцированных раковых клеток, чем в соответствующей первичной опухоли. Клетка или клетки, которые метастазируют и растут на месте метастазов, скорее всего, испытывают влияние новых микроусловий, которые могут включать и противораковые воздействия. При этом, как только клетки мигрировали на достаточное расстояние от первичной опухоли, они могут либо продолжать миграцию, либо вернуться к пролиферативному фенотипу, т.е. остаться в данном месте и начать активно делиться, в зависимости от микроокружения, с которым они столкнулись.
Важным достижением в понимании механизмов метастазирования стала концепция о предварительном формировании в отдаленных органах «ниш», привлекающих опухолевые клетки и стимулирующих размножение образующихся метастазов. Предполагается, что образование таких ниш детерминировано секрецией клетками первичной опухоли определенных цитокинов и хемокинов (плацентарный ростовой фактор, эндотелиальный фактор роста сосудов и др.), что вызывает ряд реакций в тканях некоторых отдаленных органов, приводящих, в частности, к оседанию в них VEGFR1-позитивных гемопоэтических клеток, которые, продуцируя специфический набор адгезионных молекул, протеаз и т.д. (интегрин 41, матричная металлопротеиназа 9 и др.), привлекают опухолевые клетки и стимулируют их дальнейшее размножение. «Предметастатическая» ниша была неоднократно описана на моделях животных.
Было показано, что инвазивный фенотип клеток опухоли способствовал значительной диссеминации (лат. disseminatio, от dissemino - рассеиваю, распространяю) аллотрансплантатов (орган или ткань, пересаженные между представителями одного и того же вида) у мышей. Вероятно, что большая часть мигрировавших клеток опухоли стареют или подвергаются апоптозу и лишь их небольшая часть выживает, чтобы вернуться к пролиферативному состоянию. Кроме того, частота переключения с инвазивного на пролиферативный фенотип очень низка, что объясняет ремиссию ряда злокачественных опухолей в течение длительного времени. Возможно также, что выжившие «инвазивные» клетки могут попасть в нишу, где отсутствие про-миграционных или пролиферативных сигналов заставляет их оставаться в состоянии покоя много лет, но все это время сохранять свой потенциал и при получении сигналов вернуться к пролиферативному состоянию.
Таким образом, профиль экспрессии генов, связанный с метастазированием является обратимым, что важно при разработке стратегии терапии рака.
Литература
ОСНОВНАЯ
Канцерогенез. / Заридзе Д.Г. – М.: «Медицина», 2004. – 576 с.
Онкология: Учебник для студентов медицинских вузов. / Ганцев Ш.К.. — М.: ООО «Медицинское информационное агентство», 2006. — 488 с.
ДОПОЛНИТЕЛЬНАЯ
Галицкий В. А. Канцерогенез и механизмы внутриклеточной передачи сигналов // Вопросы онкологии.- 2003.- Т.49, № 3.- С.278-293.
Б.П.Копнин Опухолевые супрессоры и мутаторные гены (avpivnik.ru/works/new/newinf05_1.doc)
Marotta L.L., Polyak K. Cancer stem cells: a model in the making// Curr Opin Genet Dev. – 2009. - 19(1). – Р. 44-50
Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. // Cell. – 2011. - 144(5). – Р. 646-674.
Molecular biology of the cell. / Alberts B. 5th ed. изд. New York: Garland Science ; [London : Taylor & Francis, distributor], 2008.
The biology of cancer. / Weinberg R. A. New York: Garland Science, 2007.
Fidler I.J. Critical determinants of metastasis. // Semin Cancer Biol. – 2002. - 12(2). – Р. 89-96.
Feinberg A.P., Ohlsson R., Henikoff S. The epigenetic progenitor origin of human cancer. // Nat Rev Genet. – 2006. - 7(1). – Р. 21-33.
Hoek K.S., Goding C.R. Cancer stem cells versus phenotype-switching in melanoma // Pigment Cell Melanoma Res. – 2010. – 23(6). – P.746-759.
Kopfstein L, Christofori G. Cancer Res. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. // Cell Mol Life Sci. – 2006. - 63(4). – Р. 449-468.
49
Основные теории возникновения и развития опухолей