Основные понятия и объекты неорганической химии
Контрольная работа
Основные понятия и объекты неорганической химии
Содержание
1. Газообразные фазы
2.Конденсированные фазы
2.1 Способ описания строения кристаллических фаз
2.2. Пределы устойчивости кристаллических структур
2.3. Дефекты твёрдого тела
2.4. Взаимодействие точечных дефектов
2.5. Жидкие фазы
2.6.Аморфные фазы.
2.7. Способы получения некристаллических твёрдых фаз
Литература
1. Газообразные фазы
Как известно, в основе кинетической теории газов лежит представление об «идеальном газе» - системе состоящей из частиц, объём которых стремится к нулю, и между которыми возможны только идеально упругие столкновения. Основные положения этой теории сформулируем в виде нескольких положений:
Газ состоит из взаимно непроницаемых молекул, заполняющих определённый объём пространства.
2. Диаметр молекул при 100 кПа составляет 1/10 расстояния между ними (т.е. суммарный объём молекул при указанном давлении приблизительно равен 1/1000 объёма системы).
3. Молекулы в газовой фазе находятся в беспрерывном движении, что предопределяет их столкновение между собой и стенками сосуда, в котором заключён газ. Эти столкновения предполагаются идеально упругими, поэтому суммарный импульс всех молекул системы в результате столкновений не убывает, несмотря на то, что при отдельных столкновениях происходит передача импульса от одной движущейся молекулы к другой.
4. Число молекул газа в единице объёма системы величина постоянная при Т = const и р = const.
5. Температура газа предопределяется средней кинетической энергией молекул, а давление в системе является усреднённой силой повторяющихся столкновений молекул со стенками сосуда.
«Идеальный газ», как система, характеризуется четырьмя параметрами состояния: объём системы (V), число молей газа в системе (n), её температура (Т) и давление (р). Используя положения кинетической теории газов, выведем уравнение состояния рассматриваемой системы. Для этого выведем функции, связывающие два параметра системы при фиксированных других параметрах.

Рис 1.
При Т = const и р = const увеличение числа молекул в системе (согласно п.4 теории газов) должно приводить к росту объёма системы, т.е. между V и n прямо пропорциональная зависимость: n = k1V или n/V = k Если теперь зафиксировать значение V, то при V = const, Т = const и р = const, величина n также будет постоянной : n = k1 const, т.е в равных объёмах газа при одинаковых внешних условиях (Т = const и р = const) содержится равное число молекул. Последний вывод есть не что иное, как закон Авогадро.
2. При n = const и р = const находим зависимость V от Т. Согласно п.5 теории газов, температура системы предопределяется кинетической энергией молекул. Рост энергии отдельных частиц увеличивает энергию столкновений между ними, что приводит к увеличению среднего расстояния между этими частицами в системе (к «расширению» системы, т.е. к росту её объёма). Тогда V = k2Т или V/Т = k2 – при неизменном давлении в системе и фиксированном числе молекул в ней отношение её объёма к температуре есть величина постоянная – закон Гей-Люссака. Более привычным выражением этого закона является форма:
Vо /То = V1 /Т1
3. При n = const и Т = const рост давления в системе уменьшает расстояние между образующими её частицами, т.е. между этими параметрами обратно пропорциональная зависимость: V = k3/р или Vр = k3 – при неизменном числе молекул системы и постоянной температуре произведение значения объёма системы на давление в ней есть величина постоянная – закон Бойля- Мариотта. Другая математическая форма данного закона: Vоро = V1р
4. При V = const и n = const, очевидно (п.5), что рост кинетической энергии молекул (т.е. температуры) будет способствовать росту силы повторяющихся столкновений молекул со стенками сосуда (т.е. давления). Тогда р = k4Т или р/Т = k4 – при фиксированом объёме системы и неизменном числе, образующих её молекул отношение давления в системе к её температуре есть величина постоянная – закон Шарля. Чаще используется другая форма этого закона: ро/То = р1/Т
Рассуждая подобным образом, можно установить и взаимосвязь между параметрами состояния системы и её кинетической энергией:
Ек=mv2 и согласно п.5 кинетическая энергия системы пропорциональна её температуре (т.е Т = k5Ек). В свою очередь значение температуры пропорциональны, как значениям объёма системы, так и значениям её давления (см. выше). Математическое правило в теории пропорциональности гласит: если одна величина одновременно пропорциональна нескольким величинам, то она пропорциональна произведению этих величин. Для рассматриваемого случая это означает: Т = k6Vр, тогда k6Vр = k5Ек или Vр = (k5/ k6)Ек, подставляя расчетные значения k5 и k6, получаем: Vр = Ек.
Используя тот же приём, найдём уравнение состояния «идеального газа»: так как значения V пропорциональны Т и n, а также обратно пропорциональны р, получаем уравнение: V = k7Тn/р или Vр = k7Тn, коэффициент пропорциональности k7 называется газовой постоянной и зачастую обозначается символом R. Тогда окончательный вид уравнения состояния «идеального газа» (уравнения Менделеева – Клапейрона): pV = nRТ. Значение R определено экспериментально путём измерения молярного объёма гелия при н.у.: R = Vр/Тn (Не – реальный газ, который наиболее близок по характеристикам к «идеальному») – (V в л – экспериментальное значение, р = 101,3 кПа, Т = 273оК, n = 1). Так как Дж = лкПа, R в этом случае равен 8,3144 Дж/мольКо. При других единицах измерения объёма и давления значение R будет отличаться от приведённого выше значения: 0,082 лат/мольКо; 62400 мм рт.ст.мл/ мольКо; 1,9872 кал/ мольКо и т.д., однако это не изменит физический смысл данной величины. Чтобы найти физический смысл R, мысленно повысим температуру 1 моля «идеального газа» на 1оК от Т до (Т + 1). Учитывая, что по условию задачи n равно единице, получаем:
рV2 = R(Т + 1) и рV1 = RТ, тогда рV2 - рV1 = R(Т + 1) - RТ или рV = R, что в соответствии с термодинамическими представлениями означает работу расширения 1 моля «идеального газа» при повышении его температуры на 1оК при р = const. Тогда физический смысл константы Больцмана (k = R/N), где N – число Авогадро – работа одной частицы газа при тех же условиях (k = 1,3810-23 Дж/частицаКо). Константа Больцмана является одной из важнейших физических величин и входит в выражения для энергии теплового движения частиц, теплоёмкости, связывает значения энтропии с вероятностью систем и т.д..
Установим теперь зависимость между среднеквадратичной скоростью частиц (v) и температурой системы:
Vр = Ек = amv2 = amv2 , где m –масса частицы, а – число частиц системы. Для простоты будем считать, что система содержит 1 моль газа, тогда а = N и pV = Nmv2, так как Nm = М (М – масса моля) получим: v2 = 3pV/М, учитывая, что для одного моля газа справедливо равенство: pV = RТ, получим: v2 = 3 RТ/М и окончательно: v = 3 RТ/М.
Подстановка в последнее уравнение значений R, Т и М показывает:
а) v при н.у. имеет значения в пределах 1500 – 7000 км/ч;
б) при прочих равных условиях скорость движения частиц газа тем выше, чем меньше значение их молярной массы.
Очевидно, что все частицы, входящие в состав системы не могут иметь ни при каких условиях одинаковые значения скоростей, так как при их столкновениях, носящих хаотический характер, происходит передача импульса от одной частицы к другой. Реальные системы характеризуются набором частиц с различной кинетической энергией, а следовательно движущихся с различными скоростями. На основе законов статистической физики Максвелл в 1860 году показал, что распределение частиц по скорости (при фиксированной температуре системы) может быть проиллюстрировано с помощью кривой распределения представленной на рис.2
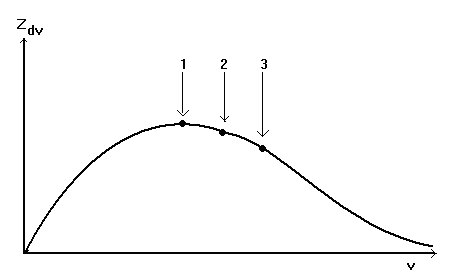
Рис.2. Распределение частиц в системе по скоростям: Zdv – число частиц, скорость которых изменяется в интервале v + dv, Z – общее число частиц, (1) - v1 – наиболее вероятная скорость, (2) - v2 – средняя скорость, (3) - v3 – среднеквадратичная скорость.
Однако, как показывают экспериментальные данные, классическая кинетическая теория газов хорошо описывает только состояние систем, образованных частицами с низкой деформируемостью, имеющими минимальный размер и минимальную энергию взаимодействия друг с другом (He, H2, Ne, Ar, O2, CO и ряд других). Но даже поведение этих газов тем больше отличается от идеального, чем ниже температура и выше давление системы. Это связано с тем, что между частицами газовой фазы существуют силы притяжения и отталкивания. При этом рост, как среднего диаметра этих частиц, так и отталкивания между ними приводят к увеличению объёма системы, по сравнению с объёмом, рассчитанным в рамках модели «идеального газа». Поэтому для согласования теоретических и экспериментальных данных в уравнении Менделеева-Клапейрона значение реального объёма (Vр.) необходимо заменить на величину (Vр.- b), которая соответствует объёму «идеального газа», с тем же числом частиц в системе. Вычитаемое (b) представляет собой поправку на увеличение объёма газа за счёт указанных выше причин и является индивидуальной величиной для молекул определённого вида ( табл. 1).
Таблица Значения поправок а и b в уравнении Менделеева-Клапейрона для некоторых веществ в газообразном состоянии.
|
молекула
|
а
|
b
|
молекула
|
а
|
b
|
|
СО2
|
3,65
|
42,8
|
HF
|
9,56
|
73,9
|
|
CS2
|
11,24
|
72,6
|
НCl
|
3,70
|
40,6
|
|
CSO
|
6,57
|
61,9
|
HBr
|
4,49
|
44,1
|
|
CSe2
|
15,62
|
91,0
|
HI
|
6,46
|
54,3
|
(а) в см6кПамоль-2; (b) в см3/моль
Очевидно, что для однотипных молекул, например, галогенов или инертных (благородных) газов с ростом объёма частиц значения (b) будут увеличиваться.
Кроме упругих столкновений между частицами, возможны и взаимодействия, приводящие к образованию ассоциатов, т.е. к уменьшению числа частиц в системе. Так как давление в системе прямо пропорционально числу находящихся в ней частиц, то процесс ассоциации снижает давление в системе, по сравнению с давлением, рассчитанным по уравнению Менделеева-Клапейрона. Это вызывает необходимость замены в этом уравнении реальных значений рр. на величину (рр + а/V2). Поправка (а/V2) учитывает влияние процесса ассоциации на изменение давления в реальной системе по сравнению с «идеальной». С учётом введённых поправок уравнение состояния реальных газов может иметь вид: (рр + а/V2)(Vр.- b) = = nRТ (уравнение Ван-дер-Ваальса).
Процесс ассоциации в зависимости от состава и строения молекул связан с различными типами межмолекулярного взаимодействия. Природа этого взаимодействия была выяснена в начале ХХ века, после того как был решен вопрос о строении атомов и молекул. Оказалось, что оно имеет электростатическую природу и являются результатом действия трех эффектов – ориентационного, индукционного и дисперсионного.
Ориентационный эффект.
Он возникает только между полярными молекулами, которые обладают собственным дипольным моментом. При этом молекулы вещества ориентируются друг по отношению к другу разноименными полюсами, и, в результате такого дипольного взаимодействия, возникает, хотя и лабильная, но все же относительно упорядоченная структура. В 1912 году Крезом было показано, что энергия ориентационного взаимодействия может быть выражена следующим соотношением:
Еор = – = –
где m – собственный дипольный момент молекулы, r – расстояние между центрами диполей, NА – число Авогадро, R – универсальная газовая постоянная, Т – абсолютная температура. Таким образом, Еор. тем больше, чем больше значение m и чем меньше величина r. При этом, с ростом температуры, Еор. уменьшается, так как усиливающееся тепловое движение нарушает взаимную ориентацию диполей. Вклад Еор в суммарное межмолекулярное взаимодействие велик для молекул, обладающих большим дипольным моментом (H2O, NH3 и т.д.).
Индукционный эффект.
Этот эффект возникает также только в том случае полярных молекул (для индивидуальных веществ), когда диполь-дипольное взаимодействие (за счет взаимной деформации) приводит к увеличению взаимодействующих молекул (смещение центров положительного и отрицательного заряда диполей). Как показал Дебай (1920) Еинд может быть рассчитана по формуле:
Еинд = – ( ) = –
) = – 
где – поляризуемость (деформируемость) молекулы.
Еинд возрастает по мере увеличения и деформируемости молекул и быстро падает с ростом расстояния между ними. В то же время Еинд от температуры не зависит, так как наведение диполей происходит при любом пространственном расположении молекул. Еинд в 10-20 раз меньше Еор. Более или менее ощутимое влияние индукционного взаимодействия наблюдается только для частиц, обладающих большой поляризуемостью. Например, вклад Еинд в суммарную энергию межмолекулярного взаимодействия для NH3 выше, чем для H2O . Это объясняется тем, что поляризуемость молекул NH3 выше, чем молекул Н2О, хотя (NH3)<(H2O).
Дисперсионный эффект.
Этот эффект реализуется при взаимодействии любых атомов, а также молекул, независимо от их строения и полярности, т.е. он является универсальным. В рамках квантовомеханических представлений дисперсионный эффект объясняется синхронизацией мгновенных диполей взаимодействующих частиц (Дебай 1920). Рассмотрим систему, состоящую из ядра и движущихся электронов. В любой момент времени такая система, вследствие несовпадения центров зарядов электронных облаков и ядра, представляет собой мгновенный диполь. Число таких диполей равно числу электронов системы. Электрическое поле мгновенных диполей атомов и молекул индуцирует мгновенные диполи в соседних частицах. Каждый из диполей будет влиять на ориентацию подобных мгновенных диполей, возникающих в близлежащих молекулах. В результате этого движение многих мгновенных диполей перестает быть независимым и становится синхронным. В результате частицы испытывают притяжение друг к другу и энергия системы снижается.
Едис = – = –
Где h – постоянная Планка, о – частота колебаний, отвечающих нулевой энергии, присущая каждому атому или молекуле при ОоК. Приближенно о= I1, тогда
Едис = – = –
Складывая все три составляющие Ван-дер-ваальсовых сил, получим:
Е = - – – = –
где К = А+В+С.
Величина вклада отдельных составляющих в суммарном эффекте зависит от состава и структуры молекул. Причем, наиболее существенными для межмолекулярного взаимодействия являются такие их свойства как полярность и поляризуемость. Чем выше полярность молекул, тем значительнее роль ориентационных сил. В то же время с ростом поляризуемости возрастает дисперсионный эффект. Индукционный эффект зависит от обоих факторов, но сам по себе имеет лишь второстепенное значение.
Суммарная энергия межмолекулярного взаимодействия очень мала по сравнению с энергией ковалентной связи (табл.2). Она зачастую, меньше энергии ковалентной связи в 10–100 раз. Температуры плавления веществ, образованных неполярными молекулами возрастают по мере увеличения деформируемости этих частиц. Например, при нормальных условиях F2, Cl2 – газы, Br2 – жидкость, J2 – твердое вещество. Учет межмолекулярных Ван-дер-ваальсовых сил необходим и в других случаях, например, при расчете теплоты плавления, сублимации и т.д.
Важно отметить, что процессу ассоциации способствует снижение температуры системы, а также рост давления в ней, т.е. параметров состояния системы способстующих, или уменьшению кинетической энергии частиц, или их сближению в пространстве.
Таблица 2. Энергия межмолекулярного взаимодействия для
молекулярных веществ и температуры их кипения (ТкипоС )
|
состав
|
(D)
|
П.
|
Эффект в ккал /моль
|
Е
|
ТкипоС
|
|
|
|
|
Eop
|
Еинд
|
Едис
|
|
|
|
H2
|
0
|
0,20
|
0
|
0
|
0,04
|
0,04
|
20,21
|
|
Ar
|
0
|
1,63
|
0
|
0
|
2,03
|
2,03
|
76
|
|
Xe
|
0
|
4,00
|
0
|
0
|
4,40
|
4,40
|
168
|
|
HCl
|
1,03
|
2,63
|
0,79
|
0,24
|
4,02
|
5,05
|
188
|
|
HBr
|
0,78
|
3,58
|
0,26
|
0,17
|
6,80
|
7,23
|
206
|
|
Hj
|
0,38
|
5,40
|
0,14
|
0,075
|
14,47
|
14,56
|
238
|
|
NH3
|
1,5
|
2,21
|
3,18
|
0,37
|
7,07
|
7,07
|
239,6
|
|
H2O
|
1,84
|
1,48
|
8,69
|
0,46
|
11,30
|
11,30
|
373
|
П. - поляризуемость
Однако необходимо учитывать, что степень ассоциации в системе может быть значительной, как при относительно низком давлении, так и при достаточно высоких температурах. Например, пары брома при 300оК и р = 20 кПа обнаруживают значительное отклонение от значений параметров предсказанных, не только на основе уравнения Менделеева – Клапейрона, но и с использованием уравнения Ван-дер-Ваальса. По данным УФ спектроскопии это связано с протекающим в системе процессом ассоциации: 2Br2  (Br2)2, равновесие которого, при указанных выше значениях параметров системы, смещено вправо. В связи с экзотермичностью этого процесса рост температуры системы способствует смещению указанного равновесия влево и выше 480оК значения параметров состояния паров брома удовлетворительно описываются с помощью уравнения Ван-дер-Ваальса. Аналогичные аномалии отмечены и для газовых фаз водорода (T < 35oK), азота, аргона, ксенона, кислорода (T < 85oK) и т.д. при давлениях > 25 кПа, которые связаны с образованием молекулярных и атомных ассоциатов – (H2)2, (N2)2, (O2)2, Ar2, Xe2.
(Br2)2, равновесие которого, при указанных выше значениях параметров системы, смещено вправо. В связи с экзотермичностью этого процесса рост температуры системы способствует смещению указанного равновесия влево и выше 480оК значения параметров состояния паров брома удовлетворительно описываются с помощью уравнения Ван-дер-Ваальса. Аналогичные аномалии отмечены и для газовых фаз водорода (T < 35oK), азота, аргона, ксенона, кислорода (T < 85oK) и т.д. при давлениях > 25 кПа, которые связаны с образованием молекулярных и атомных ассоциатов – (H2)2, (N2)2, (O2)2, Ar2, Xe2.
Достаточно распространённым заблуждением является тезис о том, что при высоких температурах не существует проблем расчета параметров состояния газообразных систем. Во-первых это утверждение бессмысленно без учёта давления в системе (например, рассчитанный по уравнению Менделеева – Клапейрона объём СО2 при 350оК и давлении 12000 кПа в три раза больше экспериментально найденного). Во-вторых, зачастую, это утверждение базируется на данных по инертным и благородным газам, а также по газообразным фазам водорода, азота, кислорода, оксида углерода (II), т.е., либо на атомных системах, либо на системах состоящих из термически стабильных молекул. Во всех других случаях температурные интервалы, в которых результаты расчета по уравнениям состояния газов и данные экспериментов хорошо согласуются, являются достаточно узкими. Это связано с тем, что на смену процессам ассоциации, описанным выше, приходят процессы диссоциации, протекающие при температурах тем более низких, чем меньше энергия связи в молекуле. Например, у газообразных галогенов при температурах выше 600оК отмечен аномально быстрый рост давления в изолированных системах, что связано со смещением вправо равновесия реакций: Г2  2Г. Однако влияние роста температуры может быть и обратным. Например, в случае газообразных галогенводородов, рассчитанные параметры давления в достаточно широком температурном интервале оказываются меньше, чем определённые экспериментально. Это связано с высокой концентрацией в системах при определённых температурах активных комплексов:
2Г. Однако влияние роста температуры может быть и обратным. Например, в случае газообразных галогенводородов, рассчитанные параметры давления в достаточно широком температурном интервале оказываются меньше, чем определённые экспериментально. Это связано с высокой концентрацией в системах при определённых температурах активных комплексов:
2НГ 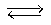 (НГ)2*
(НГ)2* 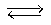 Н2 + Г2 (где (НГ)2* - активный комплекс)
Н2 + Г2 (где (НГ)2* - активный комплекс)
Таким образом, описание систем реальных газов при изменении параметров их состояния требует глубокого понимания протекающих в них процессов, приводящих к значительному изменению состава и строения входящих в них частиц. С практической точки зрения это необходимо для создания технологий, в которых газообразные фазы используются в качестве прекурсоров, или являются конечными (промежуточными) продуктами реакций. В частности, без глубокого понимания состояния газовых систем при условиях синтеза (См взаимодействующих форм, Еак. реакций, возможность изменения их механизма за счёт изменения природы реагирующих частиц и т.д.) невозможно рассчитать кинетику отдельных этапов технологического процесса и, следовательно, разработать этот процесс в целом.
2.Конденсированные фазы
Кристаллические фазы
Все твёрдые вещества можно разделить на кристаллические и аморфные, а каждый их этих типов (по природе образующих их частиц) – на атомные, молекулярные и ионные. В ряде случаев для классификации веществ используется характерный для данной системы тип связи – (ковалентные, ионные, ионно-ковалентные).
2.1 Способ описания строения кристаллических фаз
В основе большинства теорий, описывающих состояние кристаллических фаз, лежит представление об идеальном кристалле, особенностью строения которого является правильное, повторяющееся расположение в пространстве образующих его частиц. Это позволяет описывать строение всего кристалла, выделяя в нём небольшую, повторяющуюся часть, которая называется элементарной ячейкой. Элементарная ячейка представляет собой параллелепипед, построенный на трёх элементарных трансляциях. Тогда пространственную решётку можно представить как систему одинаковых параллелепипедов, которые без зазоров заполняют пространство и могут совмещаться друг с другом с помощью указанных трансляций.
Кристаллы классифицируются в соответствии с их сингонией. В сингонию объединяются кристаллы, у которых одинаковая симметрия элементарных ячеек и одинаковая кристаллографическая система осей координат (рис. 3 и 4). Общее число сингоний равно семи. К высшей сингонии относится кубическая. Это единственная сингония, которой отвечает обычная декартова система координат. К средней сингонии относятся: тригональная, тетрагональная и гексагональная, а к низшей – ромбическая, моноклинная и триклинная (рис.4).
Из законов геометрии следует, что только относительно небольшое количество геометрических форм могут при периодическом повторении полностью заполнять пространство. Как было показано Бравэ, таких геометрических форм всего 14 (решётки Бравэ). Они отличаются друг от друга значениями осей и углов, а также их соотношением. Кроме того, каждая решёта Бравэ – это группа трансляций, характеризующих расположение материальных частиц в пространстве. При выборе ячейки Бравэ используют три условия:
1) симметрия элементарной ячейки должна соответствовать наиболее высокой симметрии, к которой относится кристалл, а рёбра элементарной ячейки должны быть трансляциями решётки;
2) элементарная ячейка должна содержать максимально возможное число прямых или равных углов и равных рёбер;
3) элементарная ячейка должна иметь минимальный объём.
По характеру взаимного расположения основных трансляций или по расположению узлов все кристаллические решётки делятся на четыре типа: а) примитивные - Р; б) базоцентрированные – С; в) объёмно-центрированные – I; г) гранецентрированные – F. (рис. 3.)
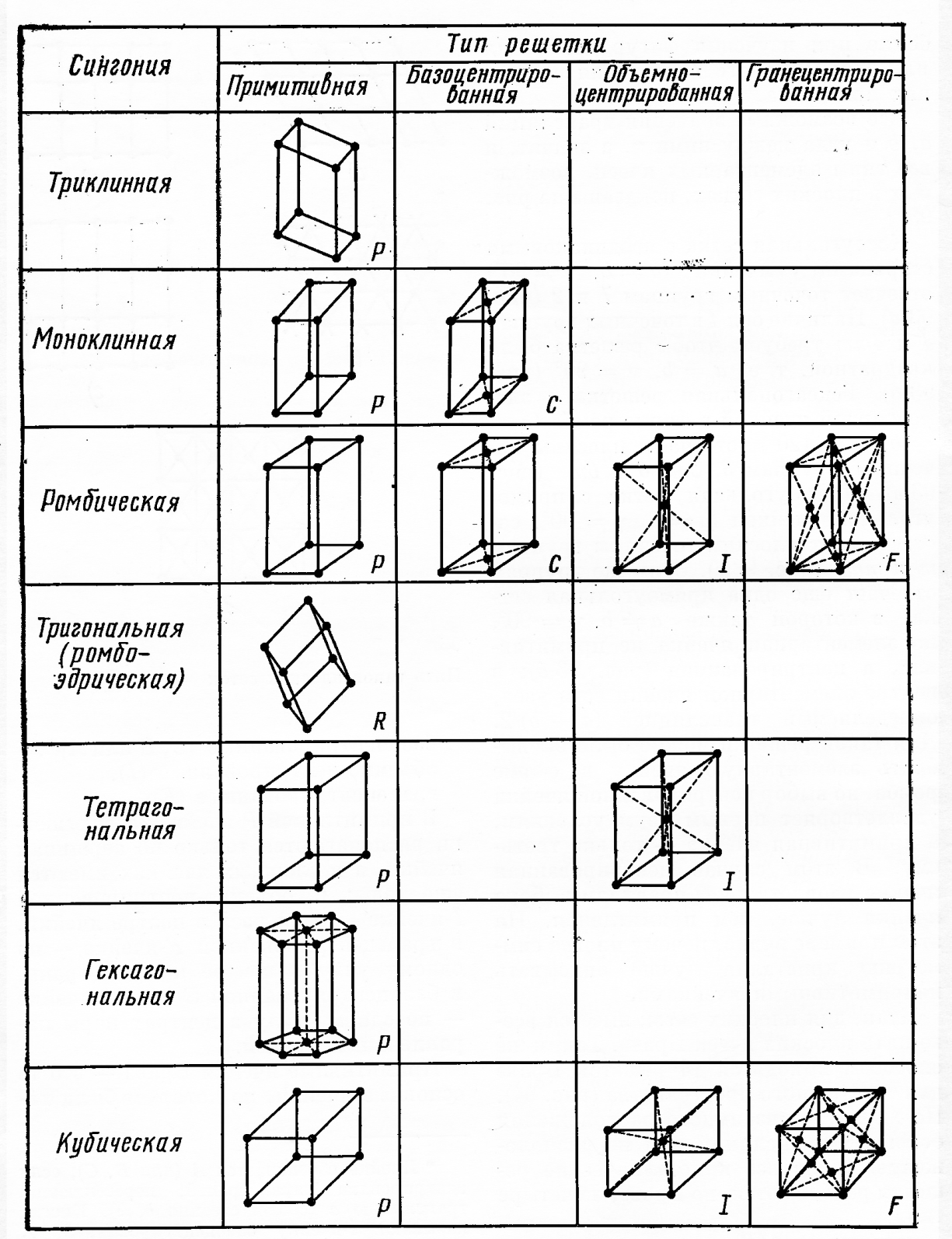
Рис. 3.Решётки Бравэ и типы сингоний
В примитивной ячейке узлы решетки располагаются только по её вершинам. В сложных ячейках кроме этого имеются еще и узлы, располагающиеся в центре ячейки (тип I), по одному в центре каждой грани (тип F) и по одному узлу в центрах пары параллельных граней (тип С) (рис. 3.)
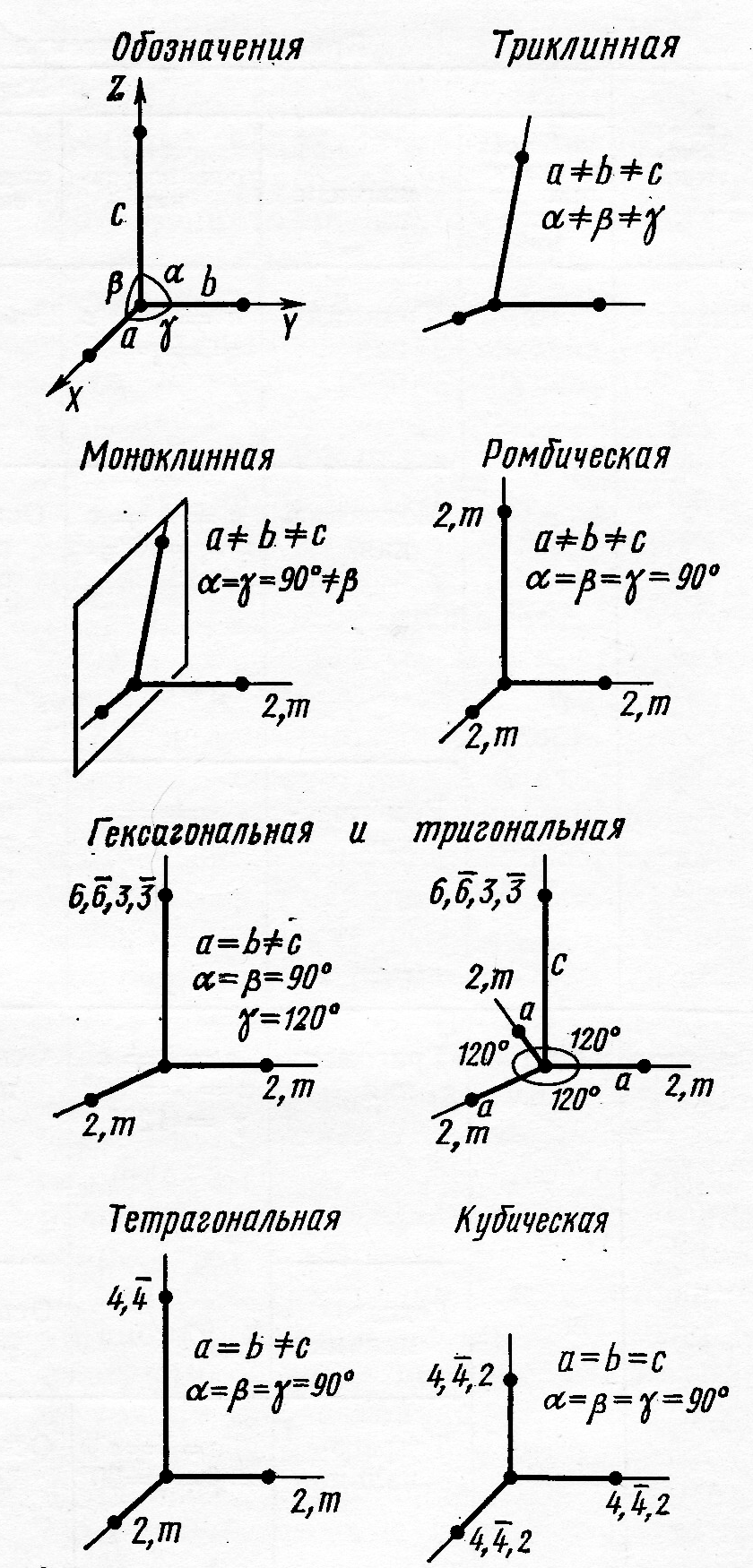
Рис.4. Кристаллографические системы координат для различных сингоний.
2.2. Пределы устойчивости кристаллических структур
Условием устойчивости любой формы (в том числе и кристалла) является минимальное (при заданных параметрах состояния системы) значение её изобарно-изотермического или изохорно-изотермического потенциалов. Одним из возможных способов достижения минимума энергии системой является максимальное сближение в пространстве её структурных единиц, т.е. их плотнейшая упаковка. Хотя тенденция к достижению плотнейшей упаковки характерна для всех типов кристаллических структур, она, практически, может быть реализована только в кристаллах с ненаправленным характером химической связи между атомами или ионами, которые в таких фазах можно считать сферическими. Таким образом модель плотнейшей упаковки предусматривает формирование структуры из частиц, имеющих сферическую симметрию и притягивающихся друг к другу, в следствии образования между ними химической связи.
В
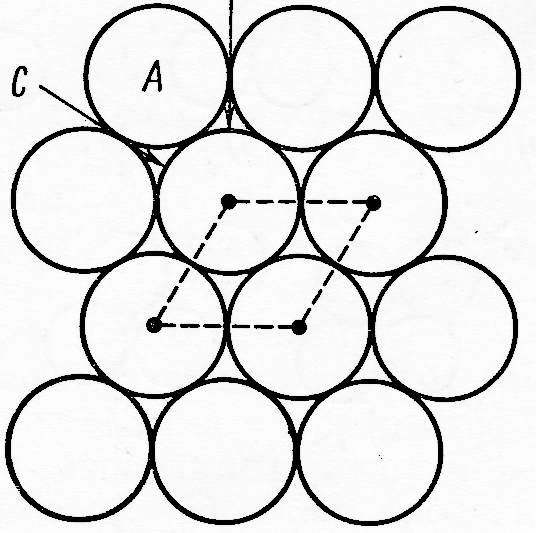
Рис.5. Однослойная плотнейшая упаковка: А – атомы, В и С – пустоты.
На рис. 5 изображён двумерный плотноупакованный слой атомов, расположенных на одной плоскости. Каждый атом такого слоя соприкасается с шестью соседними атомами и окружён шестью лунками (пустотами), а каждая из лунок окружена тремя атомами. Исходя из того, что к.ч. атомов в слое равно 6, а «к.ч.» лунок – трём, можно сделать вывод, что число лунок в слое в два раза больше чем число атомов. Для получения объёмной плотноупакованной структуры (рис.6) атомы второго слоя необходимо уложить в пустоты В или С (заполнить атомами все пустоты первого слоя невозможно с геометрической точки зрения – сравните число атомов и пустот первого слоя).

Рис.6. Двухслойная плотнейшая упаковка.
Таким образом, в двухслойной структуре возникают пустоты двух видов:
Над лункой первого слоя находиться атом второго слоя ( или лунка второго слоя над атомом первого слоя). Пустота в обоих случаях окружена четырьмя атомами, центры которых образуют правильный тетраэдр (рис.7а). Такие пустоты называют тетраэдрическими и обозначают символом Т (Т – пустоты).
2. Пустота второго слоя находиться над пустотой первого слоя и окружена шестью атомами, центры которых образуют правильный октаэдр (рис. 7б). Эти пустоты называются октаэдрическими (или О – пустотами).
Анализ двухслойной модели показывает, что число О – пустот в ней совпадает с числом атомов, образующих систему, а число Т – пустот в два раза больше числа атомов. Геометрические расчёты показывают, что условный радиус пустот типа О равен 0,41 r, а типа Т – 0,22 r, где r – металлический радиус атомов, образующих рассматриваемую кристаллическую решётку.
Задание: найдите на рисунке 6.пустоты типа Т и О.

Рис.7. Пустоты плотнейшей упаковки: (а) – тетраэдрическая, (б) - октаэдрическая
Так как во втором слое имеются два типа пустот, атомы третьего слоя можно помещать либо в лунки Т, либо в лунки О ( рис.8.).
Если атомы третьего слоя помещены в лунки Т (т.е. каждый атом третьего слоя находиться над атомом первого слоя), то третий слой повторяет укладку первого. Тогда упаковка атомов в кристалле повторяется через один слой …..АВАВАВАВ…. (рис.8а) и называется гексагональной (ГПУ – гексагональная плотнейшая упаковка). В такой структуре имеются сквозные каналы типа (О), а к.ч. частиц в ней равно 12. Если атомы третьего слоя помещены в лунки О, т.е. расположение атомов в этом слое не повторяют расположение атомов первого слоя, то тогда четвёртый слой атомов в точности расположится над первым и повторение структуры будет наблюдаться через два слоя ……..АВСАВСАВС….. (рис.8б).
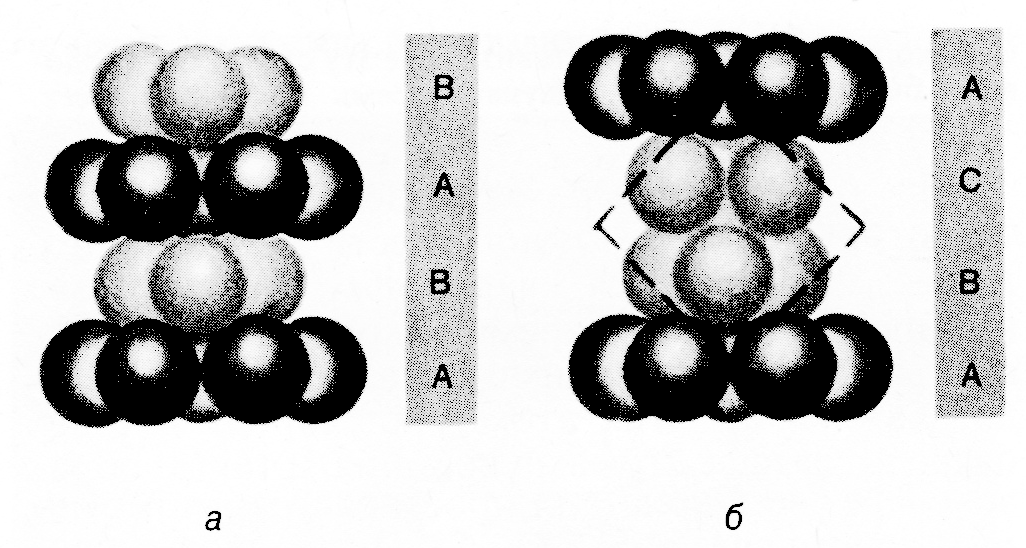
Рис.8.Гексагональная (а) и кубическая (б) плотнейшие упаковки.
Такая структура называется кубической плотнейшей упаковкой (или ГЦК - гранецентрированная кубическая структура). В отличии от ГПУ в этой структуре пустоты типа (Т) размещены над пустотами (О) и структура не имеет сплошных каналов. Общее у ГЦК и ГПУ координационное число равное 12.
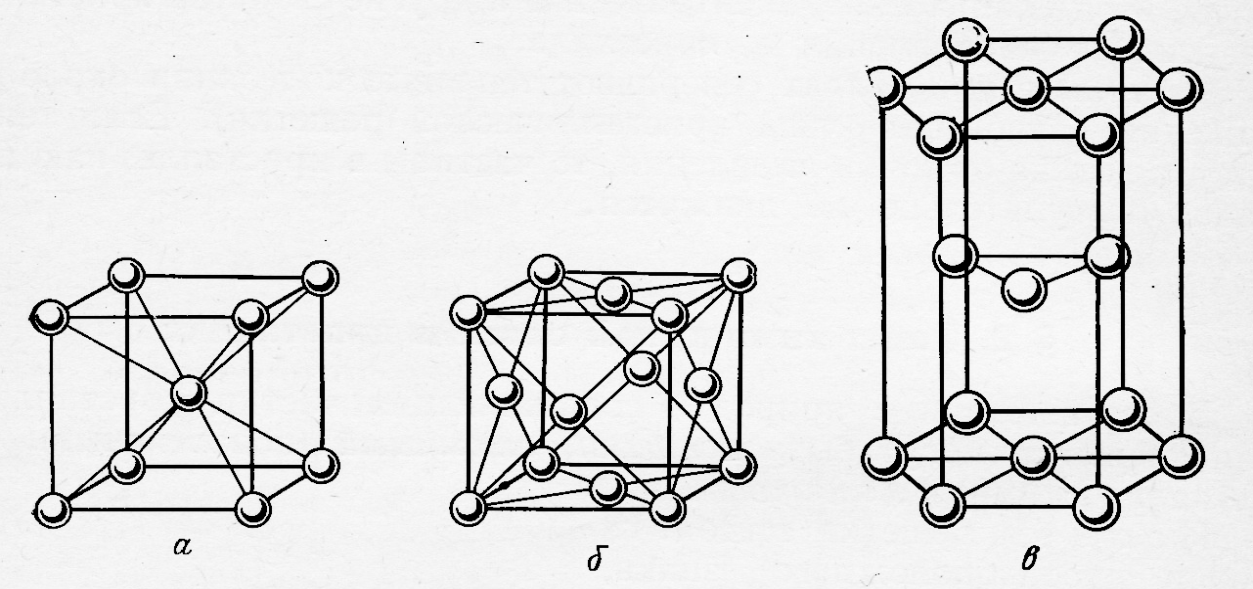
Рис.9. Элементарные ячейки структур кристаллов:
а – ОЦК, б – ГЦК, в – ГПУ.
Плотность упаковки атомов в любой структуре определяется с помощью коэффициента компактности:
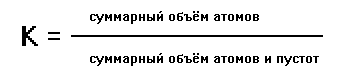
В ГПУ и ГЦК коэффициент компактности одинаков ( К = 74,05 %), т.е. атомы занимают около объёма системы.
Атомы одинакового размера можно расположить и несколько иначе (рис.10). В такой структуре атомы монослоя контактируют только с четырьмя соседними, а образующиеся пустоты являются квадратными. Атомы последующего слоя располагаются в углублениях предыдущего, порядок чередования слоёв такой же как в ГПУ, однако константа компактности в данном случае несколько меньше (К = 68 %). Структура носит название кубической объёмноцентрированной (ОЦК) и, хотя не относится к плотнейшим упаковкам, но вплотную к ним приближается.
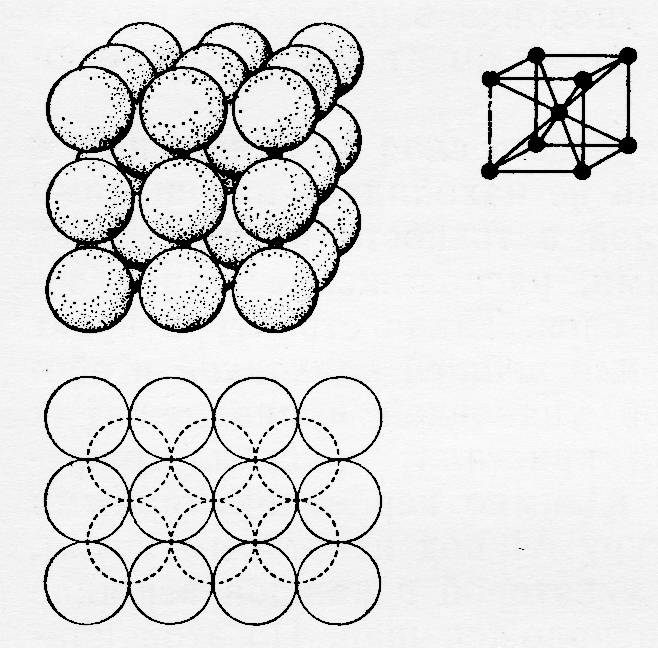
Рис.10. Объемноцентрированная структура.
Структуры, с указанным типом химической связи между частицами, будут стабильны только тогда, когда тетраэдрическая или октаэдрическая пустота будет соответствовать размерам внедряющейся в неё частицы. Рассмотрим это положение на примере ионных кристаллов (рис.11 и 12).
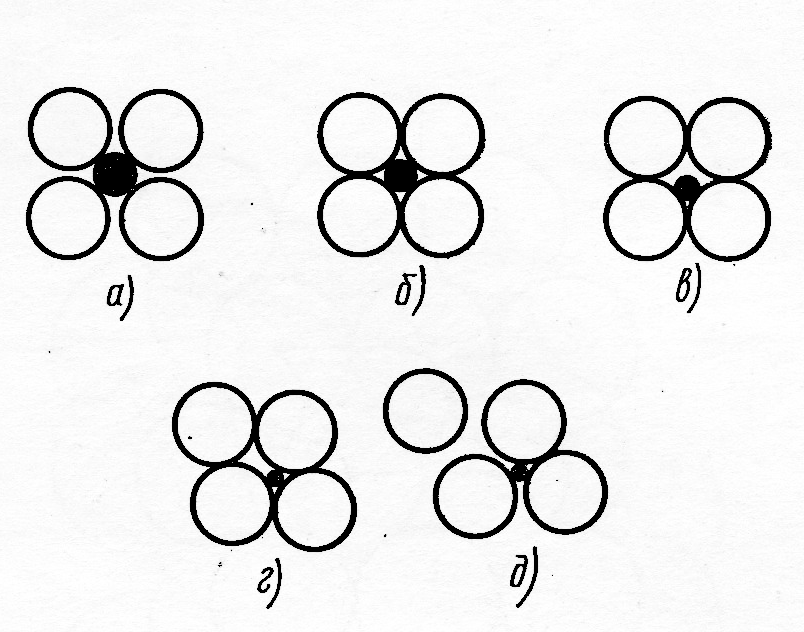
Рис.1 Изменение координации катионов при уменьшении их радиуса (не заштрихованные кружки – анионы, чёрные – катионы)
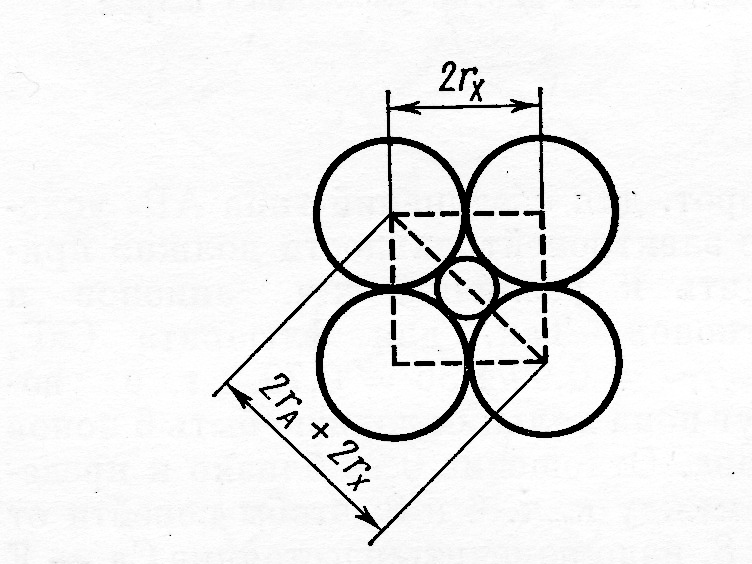
Рис.12. К расчету определения устойчивости структуры с к.ч.=6.
Из схемы, представленной на рисунке 11 следует, что вариант (а) отражает устойчивое состояние системы: каждый ион соприкасается только с противоположно заряженным ионом, а вариант (б) – предел устойчивости рассматриваемой структуры. Если продолжить уменьшать размер катиона (варианты в и г рис.11), то структура дестабилизируется за счёт роста сил отталкивания между анионами, что на определённом этапе приведёт к её перестройке и возникновению формы с меньшим к.ч. катиона (вариант д). На основе указанной схемы можно рассчитать пределы соотношений rA/rX (где А – катион и Х – анион), при которых будут устойчивы структуры с заданным координационным числом. Пример такого расчёта для структуры с к.ч.=6 иллюстрирует рисунок 12:
2(rA + rX) = 2rX2, тогда rA/rX = 2 – 1 = 0,41
Аналогичный расчёт для других типов ионных структур даёт следующие пределы их устойчивости (табл.3.1)
Таблица 3.Пределы устойчивости ионных структур.
|
rA/rX
|
к.ч.
|
координационный многогранник
|
|
0 – 0,15
|
2
|
гантель
|
|
0,15 – 0,22
|
3
|
треугольник
|
|
0,22 – 0,41
|
4
|
тетраэдр
|
|
0,41 – 0,73
|
6
|
октаэдр
|
|
0,73 – 1,00
|
8
|
куб
|
Следует отметить, что концепция плотнейшей упаковки постулирует только возможность формирования тех или иных структур при условии выполнения условия ненаправленности химической связи в кристалле. В связи с этим её механическое проецирование на структуры, например, реальных металлов не всегда оправдано. В частности, атомы элементов главной подгруппы первой группы имеют 8 (т.е. наименее деформирующуюся) предвнешнюю оболочку, что, казалось бы, в максимальной степени удовлетворяет модели плотнейшей упаковки. На практике же элементарные ячейки щелочных металлов при н.у. объёмно-центрированные т.е. не являются плотноупакованными. Другой пример – железо, которое только выше 910оС имеет структуру ГЦК, а переход этой формы в ОЦК – экзотермический. Следовательно, для процесса перехода FeОЦК в FeГЦК имеет Н>0 и, следовательно, самопроизвольно может протекать только при условии: S>0. Таким образом, для железа плотноупакованная структура с термодинамической точки зрения характеризуется большей вероятностью (большей степенью беспорядка) по сравнению с менее плотноупакованной структурой ОЦК.
Очевидно, что указанные недостатки связаны с постулируемыми ограничениями данной концепции и односторонним подходом к взаимодействию между частицами. Тем не менее эта модель (с дополнением в виде структуры ОЦК) оказалась достаточно плодотворной. Так металлические модификации простых веществ различных элементов за исключением Mn, Ga, In, Hg, Sn, Bi, Ро и Pu, действительно имеют одну из рассмотренных выше структур: ГПУ,ГЦК или ОЦК (при этом структуры Mn, Ga, In и Hg являются производными от трёх основных). При этом многие элементы имеют несколько полиморфных металлических модификаций, относительная стабильность которых зависит от параметров состояния системы. Если ограничиться только основными структурами металлов, то можно заметить что Н фазовых переходов между ними не превышает 1 кДж/моль. Другими словами, термодинамическая стабильность полиморфных металлических модификаций данного элемента близка, а какая из них будет стабильной в тех или иных условиях, предопределятся структурой энергетических зон, степенью их заселённости, энергетическими характеристиками АО, принимавших участие в формировании этих зон, а также радиусами атомов, из которых образуется рассматриваемая система. В частности, формированию у металлов структуры типа ОЦК способствует большой радиус атома элемента и наличие у него электронов только на ns подуровне (простые вещества элементов главной подгруппы I группы, а также барий и радий). В то же время литий и натрий имеют высокотемпературные модификации с плотнейшей упаковкой, а стронций и кальций высокотемпературную модификацию типа ОЦК. Металлические модификации р-элементов, за исключением Sn, Bi и Ро, имеют структуры типа ГЦК или ГПУ (идеальные или искажённые).
Кроме этого, как было показано выше, концепция плотнейшей упаковки оказалась плодотворной при описании и прогнозировании строения ионных и ионно-ковалентных кристаллов. Установлено, что в большинстве таких кристаллов, более крупные частицы одного типа образуют плотноупакованные структуры, а более мелкие – заполняют, то или иное, число пустот типа Т или О. При этом характер заполнения предопределяется отношением ионных радиусов катионов и анионов (табл. 3).
Теоретические расчёты, основанные как на взаимодействии типа притяжения в первой координационной сфере, так и отталкивания во второй были подтверждены экспериментальными данными и позволили с высокой степенью достоверности предсказывать структуры многих кристаллических фаз. Эти расчёты основаны на ранее рассмотренных пределах устойчивости отдельных структур (табл. 3) правилах, сформулированных Полингом:
1). Координационное число катиона определяется геометрическими факторами, позволяющими катиону располагаться по отношению к анионам и анионам друг относительно друга на расстояниях, обеспечивающих минимум энергии системы.
Комментарий: используя данное правило, необходимо учитывать, что критическое отношение радиусов, как критерий формирования структур работает только в том случае, когда входящие в состав кристалла частицы обладают низким (в крайне случае средним) поляризующим действием и низкой деформируемостью, а связь между ними имеет ненаправленный характер.
Для уточнения комментария рассмотрим несколько примеров:
а) Ионные радиусы Zn2+ и Fe2+ практически одинаковы (0,074 и 0,075 нм, соответственно), однако оксиды ZnO и FeO имеют различное строение. rZn2+ /rО2- = r Fe2+ /rО2- = 0,53, что согласно таблице 3соответствует к.ч.= 6. Структура FeO соответствует теоретическому прогнозу (структура типа NaCl), а ZnO имеет структуру вюрцита, в которой Zn2+ имеет к.ч.= 4. Последний факт объясняется тем, что Zn2+ имеет 18 оболочку, что в соответствии с теорией поляризации говорит о высоком поляризующем действии этого катиона и значительной деформации электронных оболочек ионов кислорода в рассматриваемой системе. Так как электронная плотность анионов стягивается к катиону, в центре полиэдра резко возрастают силы отталкивания и для их уменьшения катион вынужден уменьшить своё к.ч.. б) Аналогична ситуация возникает и при анализе структур CuCl и NaCl, в состав которых входят катионы Cu+ и Na+ с практически одинаковыми ионными радиусами (0,096 и 0,095 нм, соответственно). В CuCl ион меди с 18 оболочкой, имеет к.ч.=4, а ион натрия с 8 оболочкой в NaCl – к.ч.=6.
Представленные примеры ещё раз доказывают, что рассматриваемая концепция ограничена ионами с низким и средним значением поляризующего действия и средней и низкой деформируемостью.
Кристаллическая решётка многих ионных и ионно-ковалентных фаз может рассматриваться как совокупность повторяющихся полиэдров (тетраэдров, октаэдров и т.д.), соединённых друг с другом за счёт образования общих вершин, рёбер или граней. Способ соединения полиэдров изменяет расстояние между катионами соседних полиэдров в отношении 1: 0,58 : 0,33, что отражено во втором правиле Полинга:
2) Наличие общих рёбер и тем более граней у двух структурных полиэдров понижает устойчивость структуры.
3) Любая кристаллическая фаза, независимо от типа образующих её частиц, в целом электронейтральна.
Другими словами, в пределах ионных и ионно-ковалентных фаз осуществляется полная компенсация положительных и отрицательных зарядов частиц, входящих в состав рассматриваемой фазы.
Разработанный Полингом способ прогноза строения кристаллических фаз с преимущественно ионным характеров связи основан на понятии «сила связи», под которой понимается отношение заряда иона к его к.ч.: FC = q/к.ч., например для иона Si4+ при к.ч.=4 FC = 4/4 =1, для Al3+ при к.ч.=6 FC = 3/6 = 0,5, а для О2- при к.ч. = 3 FC = -2/3 и т.д..
Рассмотрим использование правил Полинга на примере описания ряда типов кристаллических структур.
Фазы состава КА, в которых анионы А имеют плотнейшую упаковку, а rК/rА лежит в пределах от 0,41 до 0,73. Из условий задачи следует, что катионы в данной структуре занимают октаэдрические позиции. Так как (в соответствии с концепцией плотнейшей упаковки) число октаэдрических пустот равно числу анионов, то в данном примере все они будут заняты катионами. Обозначим заряд аниона (n-), тогда его FC = n/6, FC аниона составит (-n/х), так как заряды катиона и анионы для фазы указанного состава одинаковы по абсолютной величине. Согласно третьему правилу Полинга силы связей катиона и аниона равны по абсолютному значению т.е. n/6= -n/х. Преобразуя выражение, получим х =6, т.е к.ч. аниона в рассматриваемой структуре равно шести. Эта структура называется структурой типа NaCl. Такое строение имеют галогениды всех элементов главной подгруппы I группы (за исключением низкотемпературных модификаций хлорида, бромида и иодида цезия), оксиды магния, щелочноземельных и переходных элементов состава МеО и т.д..
2. Фазы состава КА, в которых анионы А имеют плотнейшую упаковку, а rК/rА лежит в пределах от 0,22 до 0,4 Из условий задачи следует, что катионы в данной структуре занимают тетраэдрические позиции. Так как (в соответствии с концепцией плотнейшей упаковки) число тетраэдрических пустот в два раза больше числа анионов, то в данном примере катионами будет занята только половина этих пустот, т.е в структуре будут чередоваться занятые катионами и свободные полиэдры (тетраэдры). Такое расположение катионов в пространстве минимизирует силу отталкивания между ними, что с точки зрения термодинамики будет способствовать стабилизации системы. По аналогии с предыдущим примером для рассматриваемых фаз получаем: n/4= -n/х, следовательно, х = 4. Указанный тип структуры носит название структуры сфалерита. Она характерна для многих сульфидов, селенидов и теллуридов, ВеО и других фаз, удовлетворяющих условиям поставленной задачи, а также ZnO, CuCl и т.д. по причинам, описанным в комментарии к первому правилу Полинга.
3. Фазы состава КА, в которых анионы А образуют простую кубическую структуру, а rК/rА лежит в пределах от 0,73 до 1,00, что (согласно табл. 3) соответствует к.ч. катиона равному восьми. Тогда катион должен находиться в центре куба, а к.ч. аниона должно быть равным к.ч. катиона.
4. Фазы состава КА2, в которых анионы А имеют плотнейшую упаковку, а rК/rА лежит в пределах от 0,41 до 0,73. Из условий задачи следует, что катионы в данной структуре занимают октаэдрические позиции. Так как число октаэдрических пустот в данном случае в два раза больше числа катионов, то в данном примере катионами будет занята только половина этих пустот, т.е в структуре будут чередоваться занятые катионами и свободные полиэдры (октаэдры). Такое расположение катионов в пространстве минимизирует силу отталкивания между ними, что с точки зрения термодинамики будет способствовать стабилизации системы. Тогда FC катиона равна (2n/6 = n/3), а FC аниона (-n/х), тогда n/3= -n/х и х = 3. Примерами фаз с такой структурой могут служить диоксиды р- и d-элементов (TiO2, MnO2, VO2, PbO2 и т.д.).
5. Фазы состава КА2, в которых анионы А образуют простую кубическую структуру, а rК/rА лежит в пределах от 0,73 до 1,00, что (согласно табл.3.1) соответствует к.ч. катиона равному восьми. Тогда FC катиона равна (2n/8 = n/4), а FC аниона (-n/х), тогда n/4= -n/х и х = 4. Указанный тип структуры носит название структуры флюорита. Примерами фаз с такой структурой могут служить дифториды р- и d-элементов (CaF2, SrF2, BaF2, CuF2, ZnF2 и т.д.).
6. Фазы состава К2А3, в которых анионы А имеют плотнейшую упаковку, а rК/rА лежит в пределах от 0,41 до 0,73. Из условий задачи следует, что катионы в данной структуре занимают октаэдрические позиции. Так как (в соответствии с концепцией плотнейшей упаковки) число октаэдрических пустот равно числу анионов, то в данном примере общее число октаэдрических пустот в 1,5 раза больше числа катионов. Тогда FC катиона равна (3n/26 = n/4), а FC аниона (-n/х), тогда n/4= -n/х и х = 4. Указанный тип структуры носит название структуры корунда и характерен для оксидов р- и d-элементов (Al2O3, Cr2O3 и т.д.).
Как показывает практика в кристаллах с преимущественно ионным характером связи расположение анионов зачастую задано плотнейшей упаковкой, а всё разнообразие структур предопределяется вариантами расположения катионов в тетраэдрических и октаэдрических пустотах этих упаковок. В связи с этим Полинг предложил изображать структуры не виде совокупности шаров одинаковых или различных размеров, а в виде полиэдров, которые могут быть получены путём соединения центров анионов, окружающих рассматриваемый катион (рис. 13- 1). Число вершин полиэдра равно к.ч. катиона, а по расположению полиэдров в пространстве можно судить о распределении катионов в объёме системы. Модель структуры кристалла строят из слоёв полиэдров. Как было показано ранее, для плотнейших кубической и гексагональной упаковок такими полиэдрами будут тетраэдр и октаэдр, соответствующие тетраэдрическим и октаэдрическим пустотам плотнейших упаковок (рис. 13- 2а и 2b). Если рассматриваемые тетраэдры соприкасаются с другими тетраэдрами, а октаэдры с октаэдрами, то это соответствует двухслойной, т.е. гексагональной плотнейшей упаковке анионов. В случае, когда тетраэдры соприкасаются с октаэдрами, то возникает кубическая трёхслойная упаковка анионов.
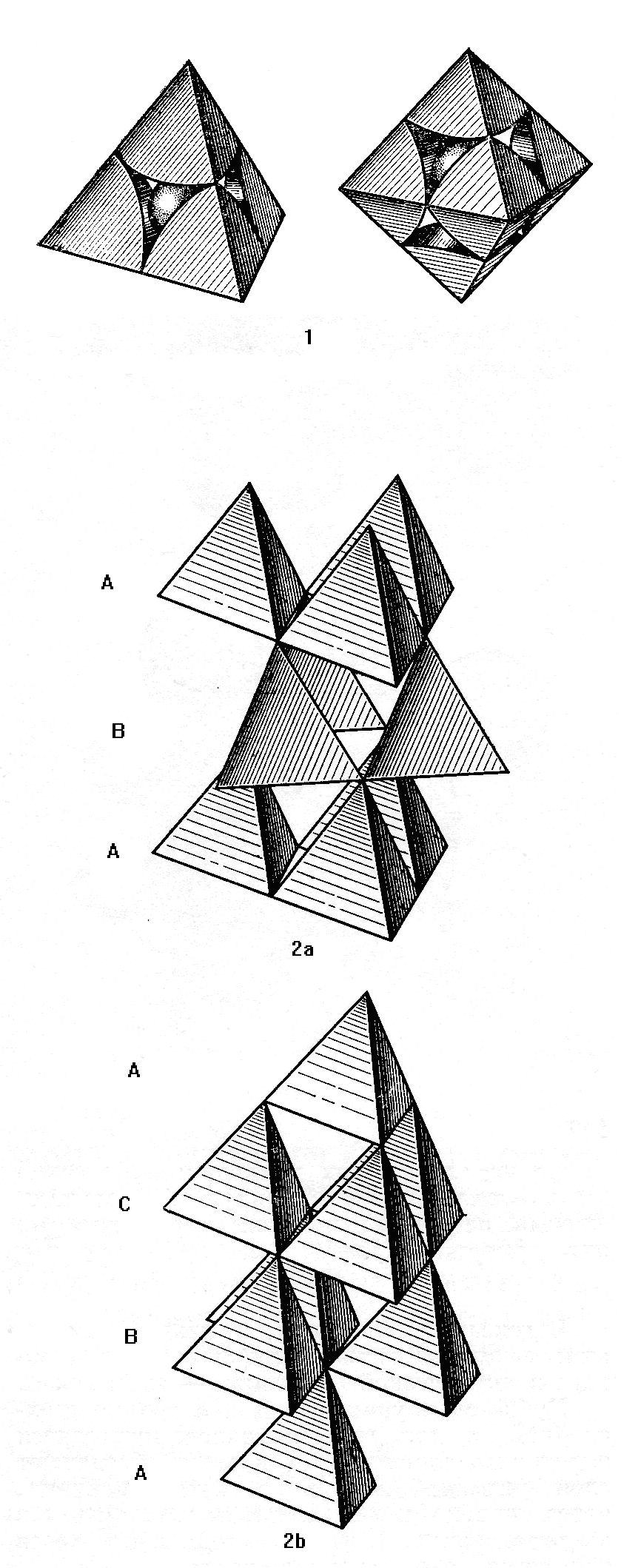
Рис. 13.- 1 – катионы внутри тетраэдрической (слева) и октаэдрической пустоты (справа); 2а - кубическая и 2b – гексагональная плотнейшие упаковки, изображённые с помощью полинговских тетраэдров.
2.3. Дефекты твёрдого тела
В изложенном выше материале в качестве основной использовалась модель идеального кристалла, который, даже теоретически, существует только при 0оК. Свойства же реальных кристаллов в значительной степени предопределяются различными нарушениями идеальной, регулярной структуры. Эти нарушения называются дефектами. Рассмотрим природу и причины возникновения дефектов в реальных кристаллах.
Электроны и дырки
В идеальном кристалле при 0оК, согласно закону распределения Ферми-Дирака, все электроны должны располагаться на минимальных по энергии разрешённых энергетических уровнях. При температурах выше 0оК за счёт тепловой энергии определённое число электронов может приобретать дополнительную энергию и переходить на более высокие энергетические уровни. Указанный переход предопределяется энергией разрешённых состояний и температурой системы. В атомных кристаллах простых веществ, таких как кремний или германий, при 0 оК все валентные электроны располагаются в валентной зоне, тогда как зона проводимости остаётся свободной. При повышении температуры часть электронов из валентной зоны перемещаются в зону проводимости, что приводит к возникновению вакантных мест в системе, которые называются положительными дырками или просто дырками. Наличие электронов в зоне проводимости и дырок вызывает возникновение у фазы нового свойства – электрической проводимости полупроводникового типа. Электронное строение ионных и ионно-ковалентных кристаллов также можно рассматривать в рамках сформулированных выше представлений. При этом необходимо учитывать, что различие между простыми веществами (ковалентный, неполярным характер связи) и рассматриваемыми фазами состоит в том, что электроны и дырки у веществ второго типа локализованы вблизи ионов в гораздо большей степени, чем, например, в структуре кремния или германия. Это предопределяет различную подвижность электронов проводимости и дырок в указанных системах (подвижность у ковалентных полупроводников на несколько порядков выше, чем у фаз ионного или ионно-ковалентного типа).
Точечные дефекты
Как отмечалось выше, идеальная кристаллическая решётка теоретически может существовать только при 0оК. Повышение температуры системы способствует те только изменению её электронного строения (см.предыдущий раздел), но и к появлению структурных дефектов в результате тепловых колебаний её составных частей кристаллической решётки. Несмотря на то, что средняя амплитуда этих колебаний по сравнению с межатомным (межионным) расстоянием невелика даже при высоких температурах, из-за флуктуации энергии в системе появляются атомы (ионы), энергия которых достаточна для перехода из регулярной позиции в междоузельное пространство. Описанный процесс, приводящий к возникновению двух видов дефектов (вакансия + внедрённый атом) называется разупорядочением по Френкелю. Следует отметить, что название «точечные» для дефектов данного типа является исторически сложившимся, так как их появление приводит к нарушению регулярности кристаллической решётки вблизи области возникновения дефекта.
При миграции из регулярной позиции частица не обязательно переходит в междоузельное пространство – второй механизм образования атомных (ионных) дефектов заключается в переходе частицы на поверхность кристалла, что равносильно достройке этой поверхности. В результате такого перехода образуется только один вид дефекта – вакансия (разупорядочение по Шотке).
Неизбежность появления точечных дефектов в кристаллах при температурах выше абсолютного нуля следует из основных понятий термодинамики. Рассмотрим этот вопрос на примере формирования в системе вакансий. Стабильное состояние любого кристалла характеризуется минимумом его энергии Гиббса:
G/n = 0 ()
(G – изменение энергии Гиббса кристалла при появлении n вакансий) при этом изменение энтальпии и энтропии кристалла могут быть выражены, соответственно, через энергию образования одной вакансии по Шотке (Еs) и изменение колебательной и конфигурационной составляющей энтропии:
H = n Еs (2.)
S = Sконфиг. + Sколеб. = k ln W + nSколеб (3.)
где k – константа Больцмана, W – вероятность вакансий для одного моля узлов решётки: W = N!/(N-n)!n!. С учётом x! = x lnx - x получаем:
ln W = N ln N – (N – n) ln(N – n) - n ln n (4.), тогда:
G = n Еs - nT Sколеб. + kT [(N – n) ln(N – n) + n ln n –N lnN ] (5.)
и с учётом (): G/n = Еs - T Sколеб + kT ln n/N – n (6.)
или: n = N exp (Sколеб/k) exp ( - Еs/RT) (7.)
Исходя из полученных данных, можно сделать вывод, что при Т > 0 значения n > 0 и мольная доля дефектов в кристалле:
n/N = A exp(-Es/RT) (8.)
где A = exp(Sколеб /k) – величина, значения которой мало зависят от температуры.
В свою очередь Sколеб можно выразить через число ближайших к вакансии атомов (z), изменивших частоту колебаний от v до v1:
Sколеб 3zk ln(v/v1).
Исходя из того, что для большинства кристаллов значения А лежат в пределах 8,5 11, а значения Еs, например, для кристаллов простых веществ составляют от 1 до 2 эВ, определим концентрацию вакансий в кристалле золота при 1000оК, используя уравнение (8) с учётом значений А=9,5 и Еs = 1,09. Расчёт показывает, что в данном случае lg(n/N) примерно равен (-5), а n/N составляет 10-5.
При образовании дефектов по Френкелю число внедрённых частиц равно числу образовавшихся вакансий: ni/N = nv/N. Тогда, используя выше приведённую методику, получим:
ni/N = nv/N = A exp(-Ed/RT) (9.)
где Ed - энергия образования пары дефектов .
В кристаллах сложных ионных веществ, помимо вышеназванных, также существуют антиструктурные дефекты обусловленные обменом позиций ионов, например для фаз типа АВ: Аа + Вв Ав + Ва. Это разупорядочение подобно образованию дефектов ранее рассмотренных типов в том смысле, что при их образовании повышается, как энтальпия системы, так и её энтропия. Это приводит к росту разупорядочения рассматриваемого вида по мере увеличения температуры системы. Для анализа дефектов обменного типа рассмотрим фазу АВ, в которой частицы А и В занимают узлы и . Общее число узлов обозначим через N, тогда N = nА + nВ, (где n – число узлов, занятых частицами определённого сорта), а долю узлов занятых «родными» частицами R и R, соответственно. Очевидно, что в идеальном кристалле R = R = При полном разупорядочении анализируемой фазы только частиц А находится в узлах , тогда R = , (минимальное значение этой величины), при этом R (в соответствии с условиями задачи), также равна . Степень разупорядочения оценивают с помощью фактора Z, который можно выразить в виде отношения: Z = (R - )2 = ( - W)2, где W – доля узлов , занятых частицами В. По аналогии с другими типами дефектов получаем:
W/R = W/ R = A exp(- Ed/2kT) (10) где Ed – энергия разупорядочения на пару частиц.
Переходы типа порядок – беспорядок наиболее часто встречаются в твёрдых растворах, например, в сплавах металлов. Это объясняется тем, что в этом случае Ed мала. В ионно-ковалентных бинарных кристаллах, напротив, эта энергия очень велика, поэтому обмен местами катиона с анионом маловероятен. В то же время в фазах более сложных по катионному (анионному) составу, Ed для катионной или анионной подрешёток может быть преодолена при температурах ниже температуры плавления фазы. В данном случае может быть достигнуто полное разупорядочение, т.к. изменение энергии кристаллической решётки в таких случаях будет обусловлено только изменениями во вторичной координации частиц, а первичная – останется неизменной. Примерами систем со 100% разупорядоченностью могут быть твёрдые растворы систем NiO – MgO и Cr2O3 – Al2O3, в состав которых входят катионы, имеющие одинаковые заряды и близкие значения ионных радиусов. Высокая степень разупорядочения наблюдается и в катионных подрешётках фаз состава Li2Fe2O4 и Li2TiO3, которые имеют структуру типа NaCl. Эти фазы, несмотря на их различный количественный и качественный состав, образуют неограниченные твёрдые растворы не только между собой, но и с NiO, MgO и другими оксидными и фторидными фазами, имеющими тот же структурный тип. В (NH4)3MoO3F3 и других оксифторидах зафиксировано разупорядочение в анионной подрешётке. Одним из признаков, по которым можно прогнозировать возможность возникновения в кристалле дефектов рассматриваемого типа, является кристаллическое строение фазы. Например, в структуре шпинели АВ2О4 ионы кислорода образуют плотнейшую кубическую упаковку. При этом число октаэдрических пустот превышает число катионов в 4/3 раза, а тетраэдрических – в 8/3 раза. Очевидно, что при близком значении ионных радиусов катионов А и В получить упорядоченную фазу указанного состава будет невозможно даже при низких температурах.
При повышении температуры системы за счёт роста энергии отдельных частиц степень разупорядочения всех фаз возрастает (рис. 14). Например, для фазы Ag2HgI4 при температурах от 0 до 176оК катионы серебра и ртути находятся в регулярных позициях. В интервале 176 - 770оК у кристаллов Ag2HgI4 происходит постепенное разупорядочение катионной подрешётки, а при Т > 770оК катионная подрешетка этой фазы полностью разупорядочена.
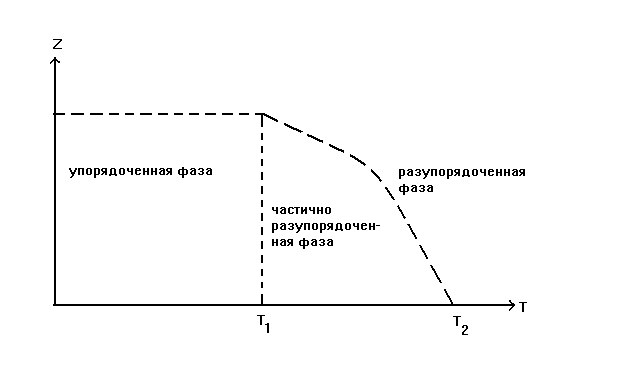
Рис.14.Изменеие степени разупорядочения фаз (Z) в зависимости от температуры системы (Т1 температура, ниже которой фаза упорядочена; Т2 – температура, выше которой фаза полностью разупорядочена.
Следует отметить, что в кристаллической решётке любого немолекулярного вещества все возможные виды точечных дефектов присутствуют одновременно, но по причине различной энергии активации процессов разупорядочения их содержание в единице объёма системы может отличаться на несколько порядков. При этом в стехиометрическом кристалле доминирует не один, а минимум два дефекта. Это следует из того, что возникновение, например, дефекта в катионной подрешётке вызывает (для сохранения стехиометрии состава или электронейтральности кристалла) образование эквивалентного числа вакансий в анионной подрешётке или эквивалентного числа внедрённых катионов, а также эквивалентного числа антиструктурных дефектов типа Ав.
В нестехиометрическом же кристалле обычно доминирует только один вид точечных дефектов. Например, в бинарном кристалле АВ, поглощающем избыток частиц В из газовой или жидкой фазы, возникают дефекты типа внедрения: В2 В или вакансий в катионной подрешётке:
В2 Вв + Vа.
Для описания процессов образования дефектов в кристаллах используется схема, основанная на представлении об окислении или восстановлении внедряемых атомов. Например, согласно экспериментальным данным кристаллы ZnO часто содержат избыток цинка в виде внедрённых атомов. Т.е. представляют собой фазу, состав которой может быть записан в виде Zn1+xO или ZnO1-x, тогда для достижения минимума энергии системой, внедрённые атомы, находящиеся в окружении отрицательно заряженных ионов кислорода должны ионизироваться, т.е. они являются донорами электронов. Сохраняя введённые ранее обозначения, процесс образования дефектов в ZnO можно выразить уравнением:
ZnZn + Oo O2 + Zn O2 + Zn2+ + 2
аналогично процесс дефектообразования в кристаллах состава Fe2+xO3:
2FeFe + 3Oo 3/2 O2 + 2Fe 3/2 O2 + 2Fe3+ + 6,
а в кристаллах UО2+x : O2 + V О О2- + h+ (где h+ - дырка).
Местом локализации дырок в решётках немолекулярных кристаллов являются обычно катионы элементов, имеющие несколько относительно стабильных степеней окисления (например, для UО2+x это U5+). В кристаллической решётке UО2+x на каждый внедрённый ион О2- образуются две дырки (два иона U5+). Таким образом, в отличие от ранее приведённых примеров, внедрённые атомы кислорода выступают в роли акцепторов, захватывая электроны и формируя в системе дырки.
В технологии функциональных материалов для изменения электрических и электрофизических свойств материалов применяется метод легирования – введение в систему примесных атомов или ионов. В процессе легирования катионы и анионы, входящие в состав добавок, занимают места в катионной (анионной) подрешётке кристалла, замещая катионы (анионы) основной фазы. Например, при легировании оксида циркония оксидом кальция имеет место следующий процесс:
ZrO2+xCaO Zr1-xCaxO2-x,
т.е. в анионной подрешётке возникают вакансии, что приводит к возникновению у материала нового свойства – ионной проводимости по ионам кислорода:
Оо O2 + 2 + Vо.
Частицы легирующих добавок (атомы или ионы) могут входить в междоузлия, при условии, что при таком размещении искажения основной решётки минимальны.
Разупорядочение кристаллов (образование вакансий, внедренных атомов и ионов, антиструктурных и примесных дефектов) часто сопровождается электронным разупорядочением (образованием электронов и дырок, свободных или локализованных на ионах, входящих в состав кристаллической решётки).
В некоторых кристаллах возможны и другие отклонения от идеальной модели. Одним из таких отклонений является колебание частиц в кристалле относительно равновесной точки при Т > 0оК. Эти упругие колебания близки к гармоническим, т.к. силы, действующие между частицами, практически точно подчиняются правилу Гука. Решения для движения частиц в решётке могут быть проквантованы, т.е. колебательная энергия изменяется только дискретно и связана с единичным квантом возбуждения упругих колебаний, который называется фононом. Каждая кристаллическая решётка имеет характерное для неё распределение фононов по разрешённым энергетическим состояниям, включая как продольные волны, или волны сжатия (сближение или удаление частиц при движении воль одной оси), так и поперечные движения (частицы движутся перпендикулярно оси). При этом соседние частицы в кристаллической решётке могут двигаться одновременно, находясь в одной фазе (акустические колебания) или в противоположной фазе (оптические колебания). Знание фононного спектра позволяет интерпретировать теплоёмкость кристаллических фаз, что помогает уточнять величины изменений термодинамических функций в ходе твёрдофазных превращений.
Другим видом дефектов в кристалле является экситон, который можно рассматривать как связанную пару (электрон + дырка), представляющую собой в целом нейтральный центр, с энергией более высокой, по сравнению с основным состоянием и перемещающеюся в твёрдом теле за счёт диффузии.
Подводя итог, можно сделать вывод, что любая твёрдая фаза характеризуется наличием различного вида равновесных электронных и точечных дефектов и концентрация каждого из них является функцией температуры, давления и состава системы.
Линейные и плоские дефекты
Реальные кристаллы кроме точечных, всегда имеют разнообразные протяжённые дефекты, располагающиеся вдоль линий и плоскостей. К ним, в частности, относятся дислокации и плоские дефекты. Характерная особенность этих дефектов заключается в том, что в отличие от точечных они не являются равновесными и, следовательно, их концентрация не может быть определена с помощью термодинамических расчётов.
Дислокации (линейные дефекты) могут быть двух видов – краевые и винтовые. Краевая дислокация – это дополнительная неполная плоскость, расположенная в кристаллической решётке, «расклинивающая» регулярные плоскости (рис.15) и распространяющаяся вдоль линии, перпендикулярной плоскости рисунка.
Как видно из рисунка, плоскости в непосредственной близости от дислокации деформированы, что помимо наличия самой дислокации повышает энергию системы. Нормальный порядок частиц в решётке, в обоих направлениях перпендикулярных дислокации, наблюдается только на расстоянии нескольких элементарных ячеек. Краевые дислокации могут при определённых условиях перемещаться в объёме кристалла – это движение называется скольжением и играет основную роль в процессе деформации кристалла.

Рис.15. Краевая дислокация

Рис.16. Движение краевой дислокации при сдвиге, приводящем к скольжению.
Необходимо отметить, что деформация кристалла не предусматривает одновременного разрыва всех связей между частицами, расположенными в соседних плоскостях – энергетически более оправданным является сдвиг за счёт движения дислокаций (рис.16).
Если краевая дислокация движется не в направлении плоскости скольжения, а перпендикулярно этой плоскости, то такой вид движения называется переползанием (рис.17).
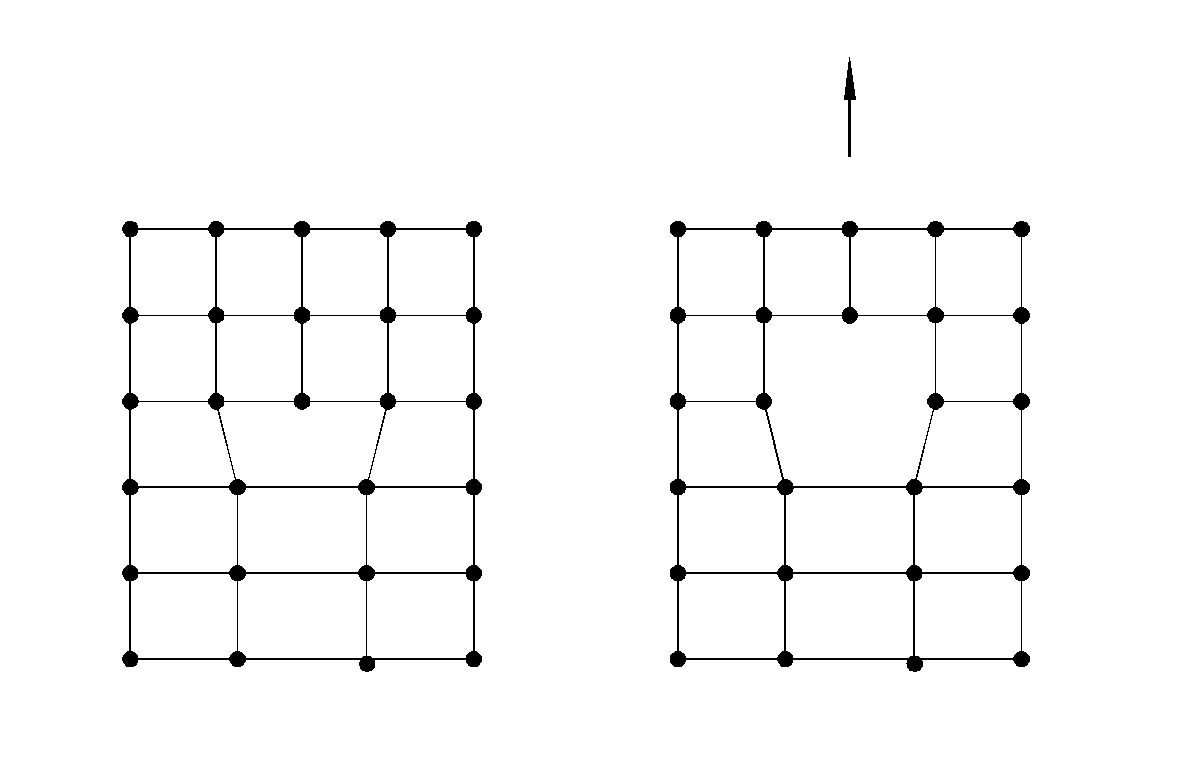
Рис.17. Движение краевой дислокации при сдвиге, приводящем к переползанию.
Результатом такого движения является образование или исчезновение вакансий или частиц в междоузлиях (в зависимости от характера движения дислокации), так как для продолжения или сокращения неполной атомной плоскости, образующей дислокацию, требуется присоединить к ней или удалить из неё определённое число частиц.
Граница между частью кристалла, в которой скольжение уже произошло, и недеформированной его частью называется линией дислокации. Линия дислокации может быть как перпендикулярной направлению скольжения (для краевой дислокации), так и параллельной направлению скольжения, тогда дислокация называется винтовой (рис. 18). Дислокации этого типа образуются в результате смещения атомов в одной части кристалла по отношению к другой его части, в результате которого вокруг линии дислокации образуется наклонная плоскость в виде спирали. В отличие от краевой дислокации, где край неполной плоскости формируется внутри кристалла, при образовании винтовой дислокации искажение элементарных ячеек происходит только в непосредственной близости от дислокации. Линейные дислокации зачастую представляют собой не прямые, а ломаные линии в виде порогов или ступенек, при этом реально существующие в кристаллах дислокации являются произвольной комбинацией краевых и винтовых.
Рис.18.Винтовая дислокация.
Плотность дислокаций в кристаллах, выращенных в равновесных условиях, порядка 102 см-2. После пластической деформации концентрация дислокаций резко возрастает (106 - 1011 см-2), что значительно повышает внутреннюю энергию системы.
Следует отметить, что многие структурные дефекты представляют собой атомные плоскости или поверхности (дефекты не обязательно должны располагаться в одной плоскости). В частности, к геометрическим поверхностным дефектам относятся поверхности и границы зёрен в керамике. Керамика представляет собой твёрдый агрегат, состоящий из кристаллических образований (зёрен), ориентированных в пространстве произвольным образом. В связи с этим слои частиц на границе зёрен также ориентированы произвольно, что означает нарушение строения кристаллической решетки, т.е. возникновение дефекта в виде поверхности произвольной формы (рис.19).

Рис.19.Границы зёрен, и области протяжённых дефектов
2.4. Взаимодействие точечных дефектов
Образование точечных дефектов в рассматриваемых системах сопровождается увеличением энтальпии и энтропии последних. Одним из конкурирующих процессов, понижающий стремление системы к максимуму энтропии должен быть процесс ассоциации дефектов. Простейшим примером прямого взаимодействия дефектов является образование ионной пары. Например, при легировании фторида кальция фторидом иттрия ионы Y3+ располагаются на позициях ионов Са2+, что приводит либо к образованию вакансий в катионной подрешётке (с учётом электронейтральности кристалла два иона Y3+ замещают три иона Са2+), либо к образованию внедрённых ионов фтора. Если принять последний механизм доминирующим, два противоположно заряженных дефекта (примесный и внедрённый ионы) притягиваются друг к другу за счёт сил электростатического взаимодействия и диффундируют навстречу друг другу. С ростом температуры равновесие указанного процесса смещается в сторону диссоциации ионной пары, но даже при высоких температурах концентрация ионных пар в кристаллах сравнима с концентрацией неассоциированных дефектов. Необходимо отметить, что сближение дефектов приводит к появлению упругих напряжений, которые могут быть уменьшены за счёт перегруппировки соседних атомов или ионов. Указанные перегруппировки предопределяют возникновение локальных нарушений порядка в микрообъёмах, окружающих дефектный комплекс.
Установлено, что взаимодействие точечных дефектов может привести и к более глубоким структурным изменениям фазы, в частности к образованию сверхструктуры или структуры сдвига. Сверхструктура возникает в процессе упорядочения вакансий или внедрённых частиц: одинаковые по заряду дефекты стремятся удалиться друг от друга на максимально возможное расстояние, что обеспечивает локальный минимум энергии системы за счёт уменьшения электростатического отталкивания между этими дефектами. Поэтому при достижении некоторой критической концентрации дефектов в кристалле, последние полностью упорядочиваются с образованием нового состояния системы. Это состояние в отличие от исходного характеризуется иным расположением частиц и вакансий в кристаллической решётке, т.е. возникает новое качество – сверхструктура. Упорядоченные дефекты менее подвижны по сравнению с не ассоциированными. Во вновь возникшем кристаллографическом порядке они уже не могут рассматриваться как дефекты: в качестве дефектов теперь будут рассматриваться любые нарушения сверхструктуры (рис.20).
Второй результат упорядочения (перегруппировка координационных полиэдров) приводит к уменьшению отношения (число анионов/число катионов) в пределах некоторых плоскостей кристалла, которые можно рассматривать как плоскости кристаллографического сдвига (рис.21). Плоскости кристаллографического сдвига являются поверхностью соприкосновения двумерных блоков кристалла, имеющих структуру отвечающей основному состоянию фазы при заданных параметрах состояния системы.
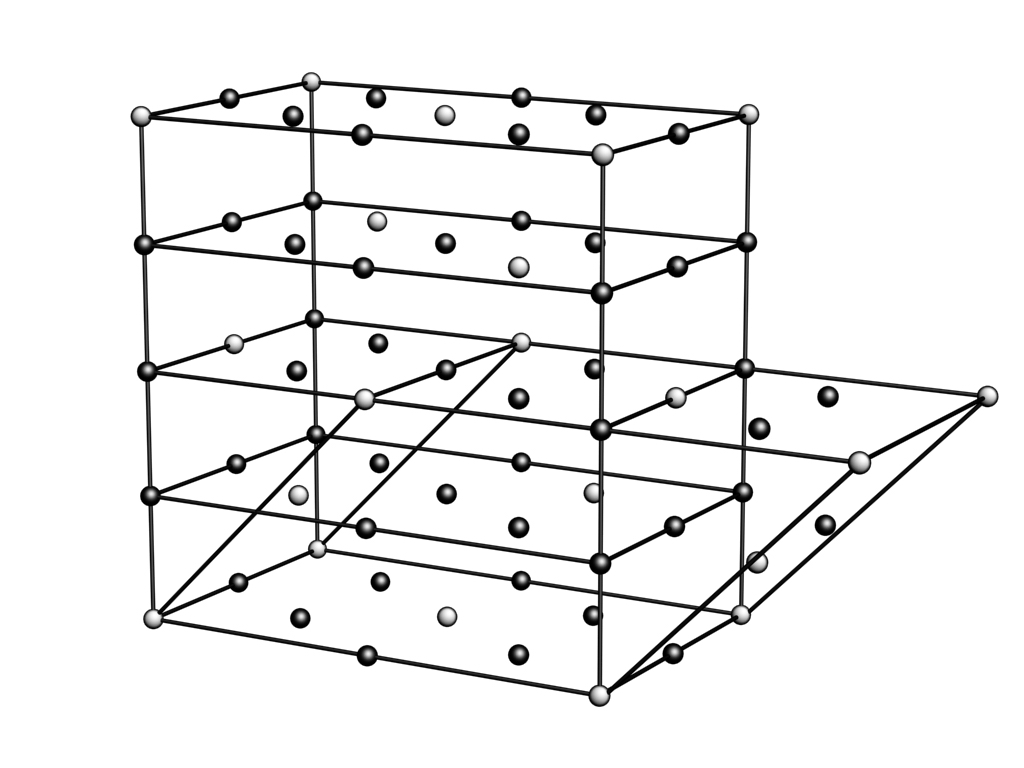
Pис. 20. Структура Fe7S8, как результат упорядочения дефектов структуры типа В8 («Fe S»).
Следовательно, в данном случае состав фазы задаётся двумя факторами: толщиной двумерных блоков с ненарушенной структурой и характером перегруппировки полиэдров в плоскости сдвига.
В настоящее время установлено, что под действием напряжения сдвига в фазах кислородно-октаэдрического типа происходит смещение октаэдров. В результате этого увеличивается число полиэдров, соединённых рёбрами, в результате чего образуются фазы типа МnO3n-1 или MenO2n-1, совокупность которых представляет собой гомологические ряды, отдельные гомологи которых отличаются частотой повторения плоскостей сдвига.
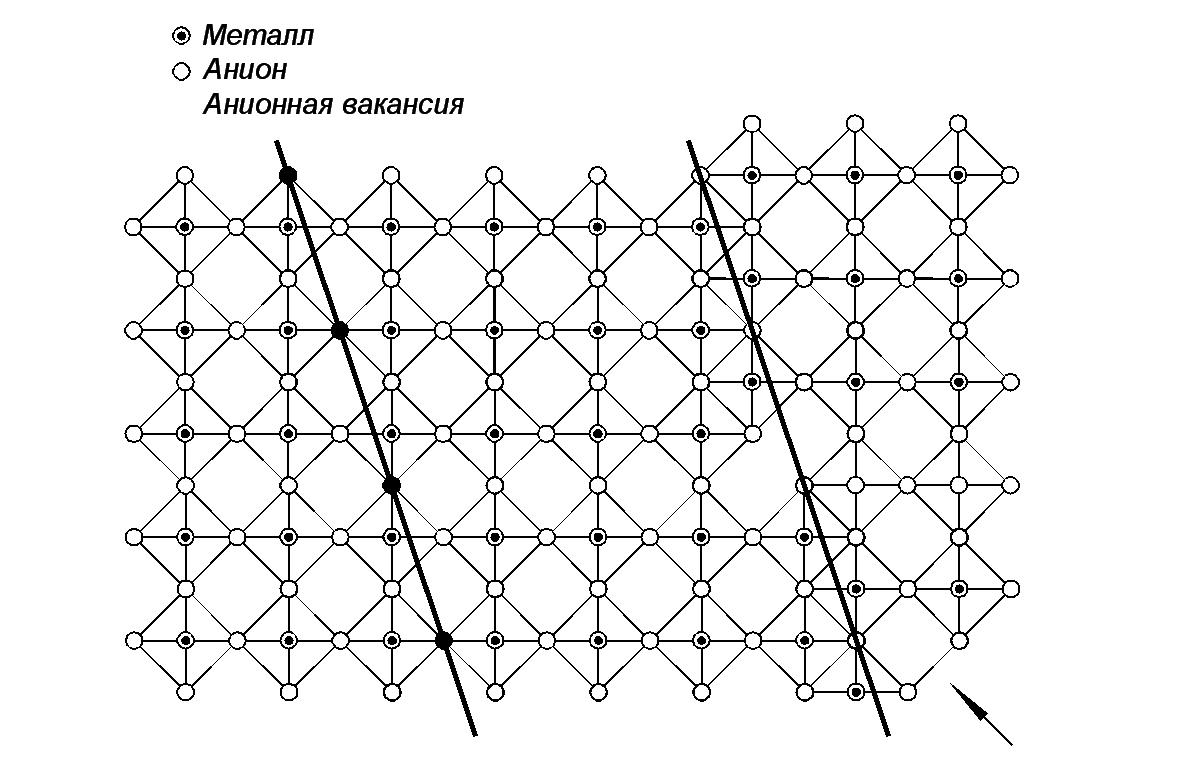
Рис.2 Результат ассоциации дефектов в структуре ReO3 – возникновение плоскости кристаллографического сдвига.
Для описания процессов формирования указанных фаз предложено несколько моделей:
Анионные вакансии, образующиеся в процессе восстановления оксидной фазы, упорядочиваются в пределах плоскостей, вдоль которых происходит кристаллографический сдвиг, связанный с аннигиляцией вакансий. При этом постулируется, что упорядочение может происходить только при высокой плотности вакансий (например, для реакции 20WO3 = W20O59 + 0,5O2 плотность вакансий должна составлять 3,7% ).
2. Также как в предыдущем случае первая стадия представляет собой возникновение беспорядочно распределённых по объёму кристалла вакансий. По мере роста их концентрации происходит процесс упорядочения и вакансии объединяются в ассоциаты, имеющие форму дисков. Контуры этих дисков представляют собой дислокации, которые за счёт действующим вдоль них напряжениям «переползают», захватывая новые вакансии. В отличии от первой модели, в данном случае для возникновения структуры сдвига не требуется высокой концентрации анионных вакансий и плоскости сдвига могут зарождаться не только на поверхности восстанавливаемой фазы, но и в любой другой части кристалла .
3. В процессе восстановления оксида с поверхности кристалла удаляется плоскость кислородных ионов (рис.22), что приводит к коллективной миграции поверхностных катионов в октаэдрические пустоты структуры, т.е. плоскость сдвига зарождается на поверхности кристалла, а последующая коллективная диффузия способствует её переходу вглубь кристалла. Если процесс восстановления продолжать, то указанный акт будет многократно повторяться, что будет увеличивать число плоскостей сдвига в единице объёма кристалла. После прекращения воздействия на фазу восстановителя на последнем этапе происходит упорядочение плоскостей сдвига. В пределе разделение кристалла на блоки одинаковой толщины, соединённые друг с другом плоскостями сдвига, при трёхмерном сдвиге эти блоки будут представлять собой параллелепипеды, состав которых принято задавать числом элементарных октаэдров вдоль осей координат.
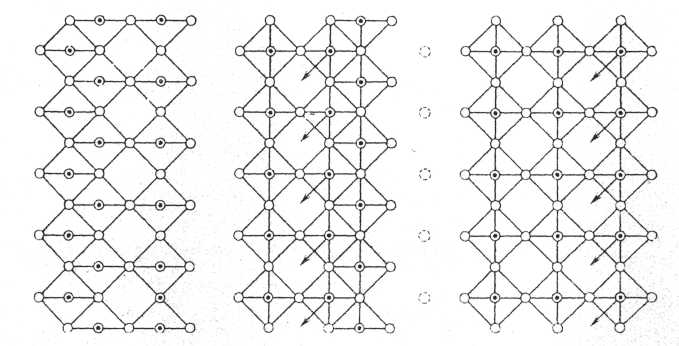
Рис.22.Образование плоскости сдвига [010] в кристаллической решётке фазы со структурой типа ReO3.
Кристаллографический сдвиг с образованием ряда фаз дискретного состава, отличающихся только частотой повторения плоскостей сдвига, не единственный способ формирования нестехиометрических фаз. Рассмотрим другие варианты этого процесса на примере структуры типа ReO3.
Структуру данной фазы (рис.22) можно представить как совокупность параллельных слоёв МеО2 (обозначим их через А) и слоёв из атомов кислорода (В): ….АВАВАВАВ….. Из выше сказанного следует, что в процессе восстановления такого кристалла, можно удалить не один, а несколько слоёв В, получив фазы с различной повторяемостью указанных слоёв ….АВААВА…., …. АВАААВА…., ….АВАА….ААВА… . При удалении двух соседних слоёв В из общего числа n, образуются фазы состава (MO2)nOn-2, что тождественно составу MnO3n-2 . Тогда при удалении трёх и четырёх соседних слоёв В получаем ряды фаз (MO2)nOn-3 (или MnO3n-3) и (MO2)nOn-4 (или MnO3n-4), соответственно. Очевидно, что указанные фазы характеризуются структурой, в которой имеются группы из нескольких октаэдров, имеющих общие рёбра.
Необходимо отметить, что кристаллографический сдвиг не является единственно возможным способом сохранения однофазности материала при его восстановлении или легировании. Перегруппировку координационных полиэдров иногда удобно представлять как процессы вращения, скольжения или отражения (рис.23 и 24).
Необходимо иметь чёткое представление о том, что механизм перегруппировки координационных полиэдров путём вращения, скольжения и отражения является одним из приёмов, позволяющим представить изменения, происходящие со структурой системы при изменении её состава. Он также позволяет установить, отличие одного родственного типа структуры от другого и не обязательно связан с глубоким преобразованием структуры. В частности, энергии активации таких перегруппировок должны быть очень высоки, что противоречит экспериментальным данным по процессам превращения одних структур в другие. Новая структурная организация как результат кристаллографического сдвига в исходной структуре возникает самопроизвольно и во многих случаях не требует больших активационных затрат.

Рис.23. Образование структуры типа тетрагональной калий вольфрамовой бронзы (б) из структуры типа ReO3 (а) путём вращения.
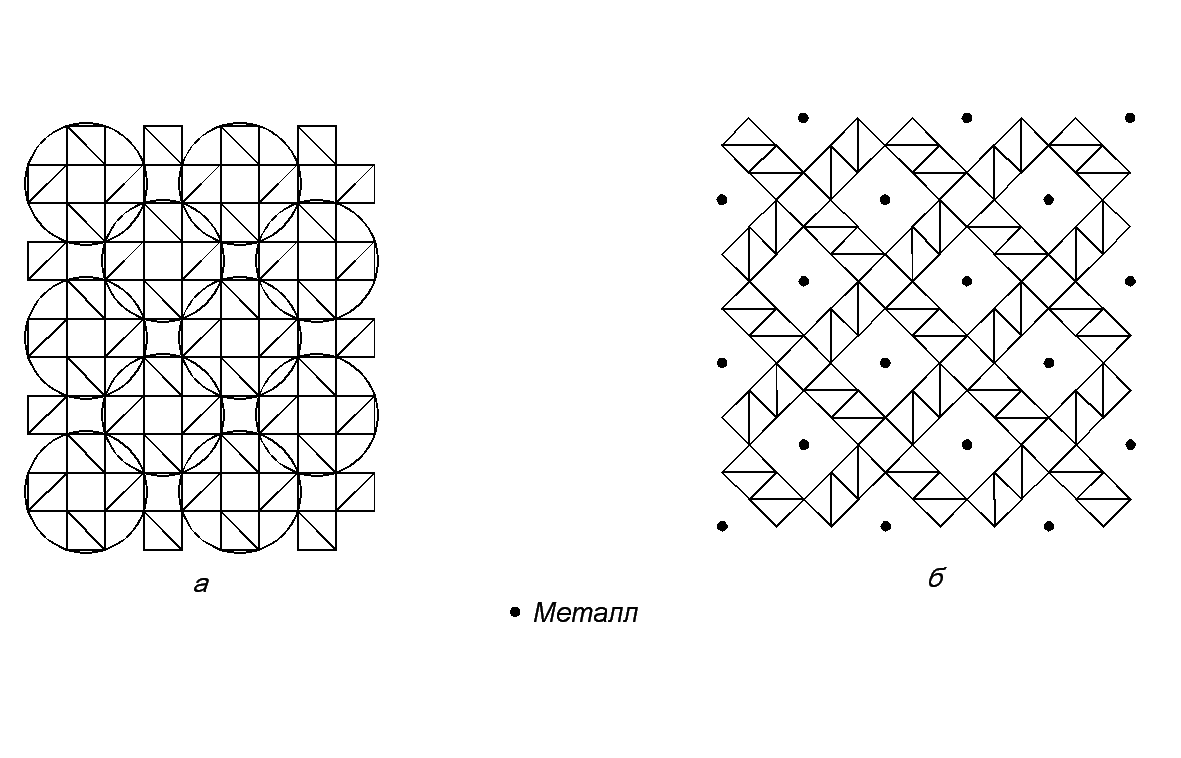
Рис. 24. Образование структуры голландита (б) из структуры рутила (а) путём вращения.
В связи с этим концепция перегруппировки, приводящей к дискретному изменению состава фаз, уже в 70-ые годы прошлого столетия была подвергнута сомнению. В частности, было установлено, что существуют системы с бесконечно адаптированной структурой, т.е. кристаллы, сохраняющие высокую структурную организацию при непрерывном изменении состава и без участия точечных дефектов. Это достигается изменением частоты повторения плоскостей кристаллографического сдвига и изменением их ориентации при сохранении толщины фрагментов мозаичной структуры. При этом первое из них происходит дискретно и приводит образованию гомологов одного ряда, а второе может осуществляться непрерывно. Комбинация обоих процессов обеспечивает непрерывное изменение состава при условии сохранения порядка. Например, бесконечно адаптированные структуры обнаружены у большого числа фаз переменного состава (Ba1+xFe2S4, TixCr1-xO1,5+0,5x, сплав церия с кадмием и т.д.). Указанные фазы, в качестве доминирующих, имеют дефекты, связанные с нарушением структурного орнамента, например, когерентное срастание участков с различной ориентацией плоскостей кристаллографического сдвига. Эти несовершенства структуры, называемые дефектами Уодсли, оказывают существенное влияние на кинетику и механизм твёрдофазных превращений.
2.5. Жидкие фазы.
Природа жидкостей пока не позволяет дать детальное описание их структуры, которое, в принципе, возможно для газов и кристаллов. Это связано с тем, что число параметров, необходимых для описания состояния газа или кристалла ограничено в первом случае, благодаря большим расстоянием между частицами, а во втором – периодическим характером кристаллической решётки.
Прежде, чем сформулировать основные положения теорий жидкости проанализируем экспериментальные данные рентгенографического и нейтронографического исследования этих объектов при температурах, близких к температурам плавления этих фаз(табл. 4 и 5).
Таблица 4. Длины связей () катион – анион и к.ч. ионов в первой координационной сфере жидкой и твёрдой фаз ионных веществ.
|
состав фаз
|
жидкая фаза
|
кристаллическая фаза
|
|
|
нм
|
к.ч.
|
нм
|
к.ч.
|
|
LiF
|
0,195
|
3,5 - 4
|
0,210
|
6
|
|
LiCl
|
0,247
|
3,5 - 4
|
0,266
|
6
|
|
NaF
|
0,230
|
4 – 4,1
|
0,240
|
6
|
|
NaCl
|
0,280
|
4 – 4,5
|
0,295
|
6
|
|
KF
|
0,270
|
4,5 – 5
|
0,280
|
6
|
|
CsI
|
0,385
|
4,5 – 5
|
0,408
|
8
|
Таблица 5. Длины связей () между одноимённо заряженными ионами и их к.ч. во второй координационной сфере жидкой и твёрдой фаз ионных веществ.
|
состав фаз
|
жидкая фаза
|
кристаллическая фаза
|
|
|
нм
|
к.ч.
|
нм
|
к.ч.
|
|
LiF
|
0,300
|
8
|
0,297
|
12
|
|
LiCl
|
0,385
|
8 - 12
|
0,376
|
12
|
|
NaF
|
0,344
|
9
|
0,339
|
12
|
|
NaCl
|
0,420
|
9
|
0,417
|
12
|
|
KF
|
0,386
|
9
|
0,396
|
12
|
|
CsI
|
0,550
|
7 – 7,5
|
0,557
|
12
|
В случае жидкостей рентгеноструктурный анализ и дифракция нейтронов не дают достаточного количества данных для характеристики распределения частиц в этих фазах. Поэтому, в настоящее время, используются формальные методы описания структуры жидкости, основанные на применении парных радиальных функций распределения, позволяющих определить вероятность нахождения пары частиц на заданном расстоянии друг от друга. В простейшем случае, когда в состав жидкости входят только два сорта частиц, выделяют три функции распределения: катион- анионную, катион – катионную и анион – анионную. Согласно полученным экспериментальным данным устойчивые флуктуации кривой радиального распределения простираются на расстояние более 1 нм, что указывает на существование чёткой тенденции к чередованию частиц в последовательных сферах координации вокруг рассматриваемой частицы. Другими словами, на основе данных эксперимента можно утверждать, что в областях жидкости с диаметром не менее 1 нм существует определённый порядок в расположении частиц, входящий в её состав. Анализируя данные таблиц 4 и 5, можно сделать ряд заключений, касающихся как структуры жидкости, так и изменения строения системы при переходе фазы от твёрдого состояния к жидкому:
Расстояние катион – анион в первой координационной сфере уменьшается за счёт снижения значений к.ч. этих частиц.
2.Расстояние между одноимённо заряженными частицами увеличивается, несмотря на уменьшение их к.ч..
Таким образом, при переходе от твёрдого состояния к жидкому, сохраняется, хотя и изменённый, ближний порядок вокруг частиц. Можно предположить, что это объясняется механизмом перехода от одного состояния к другому, связанному с образованием дефектов в исходной кристаллической решётке. Ранее (раздел 2.3) было показано, что число вакансий и дислокаций в кристалле быстро возрастает с увеличением температуры. Когда концентрация указанных дефектов достигает определённого значения, система, последовательно достигая ряда метастабильных состояний, претерпевает фазовый переход первого рода, который характеризуется скачкообразным изменением её энтропии и энтальпии.
Сравнивая значения энергии кристаллической решётки и Gпл. кристалла, можно сделать вывод, что при плавлении твёрдой фазы энергетические изменения в системе невелики и, следовательно, структурные преобразования в ней не носят глобального характера. Например, энергия кристаллической решётки NaCl составляет ~ 775 кДж/моль, а Нпл равна 28,18 кДж/моль (при Sпл.= 26,24 Дж/мольоК и Тпл. = 1074оК). Не трудно удостовериться, что Gпл. в данном случае близко к нулю. Формально можно считать, что рост энтропии при рассматриваемом фазовом переходе связан с разрывом части химических связей в системе, тогда 28/775 = 0,036, что соответствует разрыву ~ 3,6% связей в системе. Для любых атомных, молекулярных, ионных и ионно-ковалентных кристаллов отношение Нпл/Ек.р., в подавляющем большинстве случаев, не превышает 5%. С другой стороны, рост энтропии может свидетельствовать, как о возрастании подвижности частиц в системе, так и об увеличении их числа.
Следует отметить, что в ряде случаев процесс разупорядочения (в катионной или анионной подрешётках), аналогичный рассмотренному выше, наблюдается при температурах значительно более низких, чем температура плавления фазы. Например, в AgI при с.у. ионы Ag+ и I- образуют гексагональную решётку и фаза является диэлектриком. Выше 147оС электропроводность данного кристалла резко возрастает. Как показали исследования это связано с разупорядочением ионов Ag+ по отношению к ионам I-. Разупорядочение же анионной подрешётки в данном случае происходит только при 558оС, т.е. при плавлении фазы. При этом в процессе перехода AgI от твёрдого состояния к жидкому электропроводность рассматриваемой системы снижается. Интересно, что изменение энтропии при 147оС больше, чем в процессе плавления ( Sперехода = 3,5 Дж/мольоК, Sпл. = 2,70 Дж/мольоК). Аналогичное явление наблюдается и кристаллического Li2SO4 (Тперехода = 575оС, Тпл.= 850оС). В отличии от AgI в Li2SO4 зафиксировано разупорядочение в анионной подрешётке, связанное с изменением ориентации ионов SO42- ( Sперехода = 7,6 Дж/мольоК, Sпл. = 1,60 Дж/мольоК).
На основе перечисленных выше экспериментальных данных, а также представлений о решающем значении для плавления концентрации вакансий в системе, было предложено несколько моделей строения жидких фаз. Эти модели отличаются в деталях и своим математическим аппаратом, но, в целом, сводятся к следующему:
Жидкости представляют собой совокупность упорядоченных областей, размером порядка 1нм, которые называются кластерами.
2. Между кластерами существуют области пространства, в которых частицы, входящие в состав фазы, либо отсутствуют, либо их концентрация в единице объёма значительно меньше, чем в кластерах.
3. В системах присутствуют кластеры двух видов – положительно и отрицательно заряженные, суммарный же заряд системы равен нулю.
4. Частицы, входящие в состав кластеров, могут переходить от одного кластера к другому, переходя через межкластерное пространство.
5. Размеры кластеров и продолжительность их жизни уменьшаются с ростом температуры системы. При этом возрастает роль межатомных (межионных) парных взаимодействий, т.е. с ростом температуры уменьшается значение к.ч. частиц.
Указанные представления о структуре жидкости позволяют, в первом приближении, рассчитывать значения величин, характеризующие свойства жидкостей и параметры эти систем. Например, коэффициент диффузии частиц в жидкости (D):
Уравнение Стокса – Эйнштейна, выведенное на основе классических, гидродинамических представлений о движении сферы в непрерывной среде:
D=kT/6r, где – определяемый экспериментально коэффициент вязкости, r – радиус частицы. Использование этого уравнения относится к условиям описания системы через химический потенциал.
2. При описании системы через электрохимический потенциал, с учётом теории разбавленных электролитов, D определяется с помощью уравнения Нернста – Эйнштейна:
D=RT/nF2, где – экспериментально определяемая эквивалентная электропроводность; n – число молей частиц в системе; F – число Фарадея.
Расчёты, которые были проведены с помощью представленных уравнений, показали, что D частиц в жидкой фазе любого вещества в 104 – 105 раз больше, чем в кристаллических фазах того же качественного и количественного состава. Этот вывод хорошо согласуется с экспериментальными данными и ещё раз доказывает справедливость взглядов на строение жидкостей.
Говоря о свойствах ионных расплавов, следует отметить, что значения отношений Dкат./ Dанион. у них практически такие же как у кристаллических фаз вблизи температур плавления. Отсюда следует вывод, что в этих системах изменение состава кластеров в равной степени зависят, как от перемещения катионов, так и анионов.
Так же на качественном и количественном уровне теории жидкостей удовлетворительно описывают явления уменьшения площади поверхности жидкости и её смачивающие свойства. Согласно указанным представлениям, уменьшение площади поверхности жидких фаз связано с неоднородностью силового поля, оказывающего воздействие на кластеры, находящиеся на границе раздела фаз. Это связано с тем, что вектор суммарной силы (обусловленный межионным, межатомным или межмолекулярным взаимодействием) направлен к центру объёма телесного комплекса (капли). Это приводит к направленному движению кластеров от поверхности вглубь телесного комплекса, вплоть до момента равновесия указанной силы с выталкивающей силой, возникающей в приграничной области за счёт сближения кластеров, и приводит к формированию шарообразного телесного комплекса. Рассмотренный механизм реализуется в случае, когда граничащая с жидкостью конденсированная фаза не обеспечивает условий смачивания. В то же время, различное поведение жидкостей на границах с конденсированными фазами обусловлено возможностью взаимодействия частиц, находящихся на их поверхности с кластерами, входящими в состав жидкой фазы. Если энергия связей, образующихся в процессе этого взаимодействия, близка или превышает энергию связи между частицами в кластере, то хемосорбированные рассматриваемой поверхностью частицы становятся центрами упорядочения частиц жидкой фазы, т.е. жидкость смачивает поверхность конденсированной фазы. При этом, чем дальше частицы жидкости находятся от этих центров, тем меньше влияние сорбированных частиц на структуру жидкости. За счёт этого телесный комплекс принимает линзообразную форму.
2.6.Аморфные фазы.
Твёрдые вещества, не имеющие регулярной (периодической) структуры, характерной для кристаллов, носят название аморфных или некристаллических. К ним, в частности, относятся стёкла, цементы, полимерные (органические и неорганические) фазы, некоторые простые вещества, формирующиеся в условиях далёких от равновесных и т.д..
Строение рассматриваемых фаз имеет ряд общих черт со строением жидкостей:
Каждая частица, входящая в их состав, окружена примерно одним и тем же числом соседей, т.е. фиксированное к.ч. в первой координационной сфере.
2. Для второй координационной сферы к.ч. частиц данного вещества могут изменяться в некоторых пределах.
3. Расположение последующих координационных сфер рассматриваемой частицы, по мере удаления этих сфер от неё, становится всё более неопределённым.
Различия между жидкостью и некристаллической твёрдой фазой заключается в том, что время, необходимое для изменения строения жидкости, за счёт изменения параметров состояния системы или действия внешних сил (механическое напряжение, тепловые флуктуации и т.д.), мало, по сравнению с твёрдыми аморфными фазами.
К понятию аморфного твёрдого вещества можно подойти, рассматривая в качестве исходного состояния монокристалл. Как было показано ранее (раздел 3.1), любой реальный монокристалл состоит из блоков, имеющих различную ориентацию, и возникающие в монокристалле за счёт наличия в нём, например, дислокаций. Представим себе, что под воздействием сил Х объём этих блоков постоянно уменьшается до состояния, когда в блоке остаётся 1 – 2 десятка атомов. Трансляционный перенос в таких блоках на 2 – 3 межатомных (межионных, межмолекулярных) расстояния приведёт к тому, что мы переместимся из одного блока в другой, беспорядочно и случайно ориентированный по отношению к первому – т.е. указанные действия позволят осуществить переход от кристаллической к аморфной фазе.
Показанные примеры позволяют сделать вывод, что к понятию некристаллических веществ можно придти на основе представлений о не релаксирующей жидкости или с точки зрения кристалла, не имеющего дальнего порядка. Данный анализ показывает, что концепции подхода к описанию аморфных фаз не противоречат друг другу, а их применение в том или ином случае обусловлено способами указанных фаз, а именно, исходным состоянием прекурсоров.
Образование некристаллических твёрдых фаз характерно и для веществ, образованных крупными молекулами, или структура которых представляет собой совокупность цепочечных фрагментов. В таких объектах энергия связи в молекуле или цепочке заметно превышает энергию межмолекулярного или межцепочечного взаимодействия и недостаточна для формирования регулярной пространственной или плоской структуры.
2.7. Способы получения некристаллических твёрдых фаз
Известно много вариантов процессов, приводящих к получению некристаллических фаз, которые основаны на трёх основных принципах:
Конденсация парообразной фазы заданного состава при высокой скорости охлаждения. Основанные на этом принципе методы наиболее эффективны для получения аморфных фаз любого состава, однако в случае тугоплавких они сталкиваются со значительными экспериментальными трудностями.
2. Разложение гелеобразных фаз при кратковременном термическом ударе:
SiCl4 + zH2O SiO2(z-2)H2O + 4HCl SiO2(амор.)+ (z-2)H2O + 4HCl
или при их низкотемпературной частичной дегидратации, сопровождающейся образованием водородных связей:
-Al2O3 + 6H3PO4 2Al(H2PO4)3 + 3H2O смесь гидратных форм фосфата алюминия.
3. Затвердевание расплава при температурах значительно более низких, чем температура кристаллизации фазы, приводящее к образованию стекла.
Стекло представляет собой твёрдое тело, образующиеся в процессе быстрого охлаждения расплава, в ходе которого не наблюдается скачкообразного изменения свойств фазы и значений её термодинамических функций при какой – либо определённой температуре. По мере снижения температуры в таких системах, вязкость фаз в них постепенно увеличивается, что способствует уменьшению их пластичности и увеличению твёрдости.
Стеклофазу могут образовывать соединения практически всех типов: оксиды, сульфиды, соли различных кислот, простые вещества, органические соединения гидроксильного типа и т.д.. Однако известно, что стеклофазы при охлаждении жидкости могут образовывать далеко не все представители указанных групп соединений. Как было показано Захариансеном, для формирования стекла необходимо, чтобы, при фиксированном составе вещества, первые координационные числа частиц в его кристаллической и аморфной фазах были бы практически одинаковы. В то же время, изменения во второй координационной сфере не должны существенно влиять на суммарную энергию системы. Например, для стёкол на основе оксидных фаз Захариансеном были сформулированы следующие правила – для образования стеклофазы:
каждый ион кислорода должен быть связан не более чем с двумя катионами;
2. координационные полиэдры связаны между собой только вершинами с образованием трёхмерной пространственной структуры;
3. к.ч. катиона не должно быть больше четырёх;
4. каждый полиэдр должен иметь минимум три общие вершины с соседними полиэдрами.
Для уточнения отметим, что в сложных по составу стёклах (например, силикатных или боратных) только один из катионов (соответственно кремния и бора) образует с кислородом указанный тип структуры, а другие катионы располагаются в пустотах этой решётки. С целью количественной интерпретации сформулированных выше положений, Сан ввёл понятие единичной энергии связи (Е1), которая равна энергии диссоциации одного моля фазы на атомы, делённой на к.ч. катиона: Е1 = Ед./к.ч..
Для катионов, со средним значением поляризующего действия, Е1 имеет значение больше 320 кДж/моль. Как показывают эксперименты, именно оксиды этих элементов способны образовывать однокомпонентные стёкла (B3+, Si4+, Ge4+,Al3+, P5+, As3+, As5+, Sb3+, Sb5+, Zr4+ и т.д.). Для большинства указанных катионов к.ч. в стёклах равно четырём, для B3+ возможно к.ч.=3, а для Zr4+ - шести.
Если же Е1 имеет значения от 250 до 320 кДж/моль, то образование однокомпонентной стеклофазы возможно только при достаточно быстром охлаждении расплава, а её стабильность зависит от состава анионной подрешётки. Например, BeF2 легко образует стеклофазу, BeО – при очень высокой скорости охлаждения расплава, а стеклофаза BeS – не получена. К указанной группе относятся катионы: Ti4+, Zn2+, Pb2+, Al3+(к.ч.= 6), Be2+, Zr4+(к.ч.= 8), Cd2+ и другие.
Ионы же с низким поляризующим действием могут входить только в пустоты трёхмерного каркаса, образованного перечисленными выше катионами и анионами различного состава. В этих же пустотах могут размещаться и низко зарядные катионы, имеющие среднее значение поляризующего действия.
Катионы с очень высоким значением поляризующего действия (N5+, S6+, Cl7+ и т.д.) образуют молекулярные вещества, которые в ряде случаев также могут быть получены в виде аморфных фаз. Эти фазы характеризуются нарушением регулярного строения молекулярных кристаллических решёток, т.е. их строение регламентируюется другим типом взаимодействия между частицами. Так как межмолекулярное взаимодействие часто характеризуется низкой энергией, для веществ данного типа актуальна противоположная задача – определение условий способных обеспечить формирование молекулярных кристаллических фаз.
Литература
1. Бабкина, С.С. Общая и неорганическая химия. Лабораторный практикум: Учебное пособие для бакалавров и специалистов / С.С. Бабкина, Р.И. Росин, Л.Д. Томина. - М.: Юрайт, 2012. - 481 c.
2. Барагузина, В.В. Общая и неорганическая химия: Учебное пособие / В.В. Барагузина, И.В. Богомолова, Е.В. Федоренко. - М.: ИЦ РИОР, 2013. - 272 c.
3. Богомолова, И.В. Неорганическая химия: Учебное пособие / И.В. Богомолова. - М.: Альфа-М, НИЦ ИНФРА-М, 2013. - 336 c.
4. Боровлев, И.В. Органическая химия: термины и основные реакции / И.В. Боровлев. - М.: БИНОМ. ЛЗ, 2012. - 359 c.
5. Гаршин, А.П. Общая и неорганическая химия в схемах, рисунках, таблицах, химических реациях: Учебное пособие / А.П. Гаршин. - СПб.: Питер, 2013. - 288 c.
6. Грандберг, И.И. Органическая химия: Учебник для бакалавров / И.И. Грандберг, Н.Л. Нам. - М.: Юрайт, 2013. - 608 c.
7. Грандберг, И.И. Органическая химия: Учебник для бакалавров / И.И. Грандберг, Н.Л. Нам. - М.: Юрайт, 2013. - 608 c.
8. Грибанова, О.В. Общая и неорганическая химия: Учебное пособие / О.В. Грибанова. - Рн/Д: Феникс, 2013. - 249 c.
9. Захарова, Т.Н. Органическая химия: Учебник для студ. учреждений сред. проф. образования / Т.Н. Захарова, Н.А. Головлева. - М.: ИЦ Академия, 2012. - 400 c.
10. Князев, Д.А. Неорганическая химия: Учебник для бакалавров / Д.А. Князев, С.Н. Смарыгин. - М.: Юрайт, 2012. - 592 c.
11. Ливанцов, М.В. Органическая химия. Задачи по общему курсу с решениями. В 2-х т. Т. 2. Органическая химия. Задачи по общему курсу с решениями. Часть 2: Учебное пособие / М.В. Ливанцов. - М.: БИНОМ. ЛЗ, 2012. - 714 c.
12. Ливанцов, М.В. Органическая химия. Задачи по общему курсу с решениями. В 2-х т.Т. 1. Органическая химия. Задачи по общему курсу с решениями. Часть 1: Учебное пособие / М.В. Ливанцов. - М.: БИНОМ. ЛЗ, 2012. - 255 c.
13. Матусевич, Л.Г. Органическая химия. Основной курс.: Учебник / А.Э. Щербина, Л.Г. Матусевич; Под ред. А.Э. Щербина. - М.: НИЦ ИНФРА-М, Нов. знание, 2013. - 808 c.
14. Павлов, Н.Н. Общая и неорганическая химия: Учебник / Н.Н. Павлов. - СПб.: Лань, 2011. - 496 c.
15. Петров, А.А. Органическая химия: Учебник для вузов / А.А. Петров, Х.В. Бальян, А.Т. Трощенко; Под ред. М.Д. Стадничук. - М.: Изд. Альянс, 2012. - 624 c.
16. Солдатенков, А.Т. Пестициды и регуляторы роста: прикладная органическая химия / А.Т. Солдатенков, Н.М. Колядина, А. Ле Туан. - М.: БИНОМ. ЛЗ, 2013. - 223 c.
17. Титце, Л. Препаративная органическая химия: Реакции и синтезы в практикуме органической химии и научно-исследовательской лаборатории / Л. Титце. - М.: Мир, 2013. - 704 c.
18. Травень, В.Ф. Органическая химия. В 3-х т. Т. 1.: Учебное пособие для вузов / В.Ф. Травень. - М.: БИНОМ. ЛЗ, 2013. - 368 c.
19. Травень, В.Ф. Органическая химия. В 3-х т. Т. 2: Учебное пособие для вузов / В.Ф. Травень. - М.: БИНОМ. ЛЗ, 2013. - 517 c.
20. Травень, В.Ф. Органическая химия. В 3-х т. Т. 3.: Учебное пособие для вузов / В.Ф. Травень. - М.: БИНОМ. ЛЗ, 2013. - 388 c.
21. Цветков, Л.А. Органическая химия. 10-11 класс: Учебник / Л.А. Цветков. - М.: ВЛАДОС, 2011. - 271 c.
22. Шабаров, Ю.С. Органическая химия: Учебник / Ю.С. Шабаров. - СПб.: Лань, 2011. - 848 c.
Основные понятия и объекты неорганической химии