Генеалогия неорганических фаз
Контрольная работа
Генеалогия неорганических фаз
Содержание
1. Простые вещества
1.1. Способы получения простых веществ
1.2. Состав и строение простых веществ
2. Бинарные соединения
2.1. Твёрдые растворы металлического типа
2.2. Интерметаллические соединения
2.2.1. Соединения Курнакова
2.2.2. Фазы Лавеса
2.2. Соединения Юм – Розери
2.2.4. Фазы внедрения
2.3 Галогениды, оксиды и сульфиды
3 Гидроксиды и неорганические полимеры
3.1. Обзор строения и свойств гидроксидов различных элементов
3.2. Способы синтеза фаз с условной формулой ЭO2xH2O
3.3 Строение и свойства гидроксидов титана, циркония и олова (IV)
3.4. Ионообменные свойства ЭO2xH2O
3.5. Неорганические полимеры.
Литература
1. Простые вещества
Все простые вещества можно разделить на молекулярные и атомные. Первые - состоят из молекул (H2, N2, O2, O3, P4, Li2, Au2 и т.д.), связанных между собой силами межмолекулярного взаимодействия. Вторые - из атомов, между которыми может быть различный характер связи: а) дисперсионного типа (в таких простых веществах как гелий, неон, аргон и т.д.), б) ковалентный локализованный (алмаз, кремний, бор) и в) ковалентный делокализованный (в простых веществах металлического типа). При этом в парообразном состоянии существуют только аллотропные формы, образованные отдельными атомами и молекулами, а в конденсированном состоянии возможно существование модификаций любого строения. Необходимо отметить, что в ряде случаев ту или иную форму трудно однозначно отнести к одному из названных выше типов. Например, пластическая сера, красный фосфор и другие простые вещества состоят из макромолекул, образованных различным числом атомов. Макромолекулы в этих веществах ориентированны хаотично, т.е. указанные фазы относятся к аморфным. Связь между этими молекулами -дисперсионного типа, а между атомами в макромолекулах – локализованная, ковалентная неполярная. В то же время в графите, чёрном фосфоре, сером мышьяке и т.д. макромолекулы полностью упорядочены, при этом межмолекулярное взаимодействие имеет признаки делокализации, что, в частности предопределяет электронную проводимость (металлического или полупроводникового типа) этих кристаллических фаз.
1.1. Способы получения простых веществ.
Химические способы получения простых веществ можно разделить на три основных вида: восстановление, окисление или разложение прекурсоров. Восстановление различных форм сложных веществ проводят в том случае, когда целью процесса является получение простого вещества менее электроотрицательного элемента, входящего в состав прекурсора. Процессы окисления сложных веществ эффективны при получении простых веществ более электроотрицательных элементов, входящих в состав исходной фазы. Способы, основанные на термическом или электрохимическом разложении фаз, позволяют синтезировать простые вещества элементов различной электроотрицательности. В ряде случаев используются методы, основанные на физических или физико-химических свойствах простых веществ, входящих в состав смесей или растворов. Например, азот, кислород, инертные и благородные газы получают криогенной перегонкой воздуха.
Не зависимо от типа процесса, лежащего в основе способа получения простого вещества, его использование регламентируется термодинамической возможностью и полнотой протекания при выбранных условиях, а также его кинетикой (скорость, механизм, возможность подавления параллельных реакций). С учётом условий проведения процессов они подразделяются на пиро- и сольвометоды. Первые проводят при повышенной температуре, а вторые – в присутствии растворителей.
В качестве примера рассмотрим получение металлов в процессах восстановления оксидов распространёнными восстановителями, такими С и СО:
1. MexOy + yC = xMe + yCO
Go = yGoCO - GoMexOy
2. MexOy + y/2C = xMe + y/2CO2
Go = y/2GoCO2 - GoMexOy
MexOy + yCO = xMe + yCO2
Go = yGoCO2 - GoMexOy - yGoCO
Возможность получения металла из заданной формы оксида при использовании того или иного восстановителя удобно оценивать с помощью энергетических диаграмм, таких как представлена на рисунке 1.
Из данной диаграммы видно, что Al2O3 не может быть восстановлен до металла ни с помощью углерода, ни с помощью СО вплоть до температуры 2500оС, так как прямые на диаграмме, относящиеся к Go процессов окисления восстановителей, при всех рассматриваемых температурах лежат выше, чем прямая, описывающая изменение Go реакции окисления алюминия. Это означает, что Go любой из реакций восстановления (1, 2 или 3, если Ме = Al) при любой из температур рассматриваемого интервала, будет значительно выше нуля, т.е. выход продуктов реакции будет ничтожно мал.
В то же время Cu2O с термодинамической точки зрения может быть восстановлен любым из представленных восстановителей и, следовательно, конкретный выбор одного из них будет предопределяться кинетическими или технологическими критериями. Также, очевидно, что «VO» может быть восстановлен только углеродом при Т > 1700оС, при этом углерод будет окислен до СО: «VO» + С = V + СО.
- Go298 ккал/моль
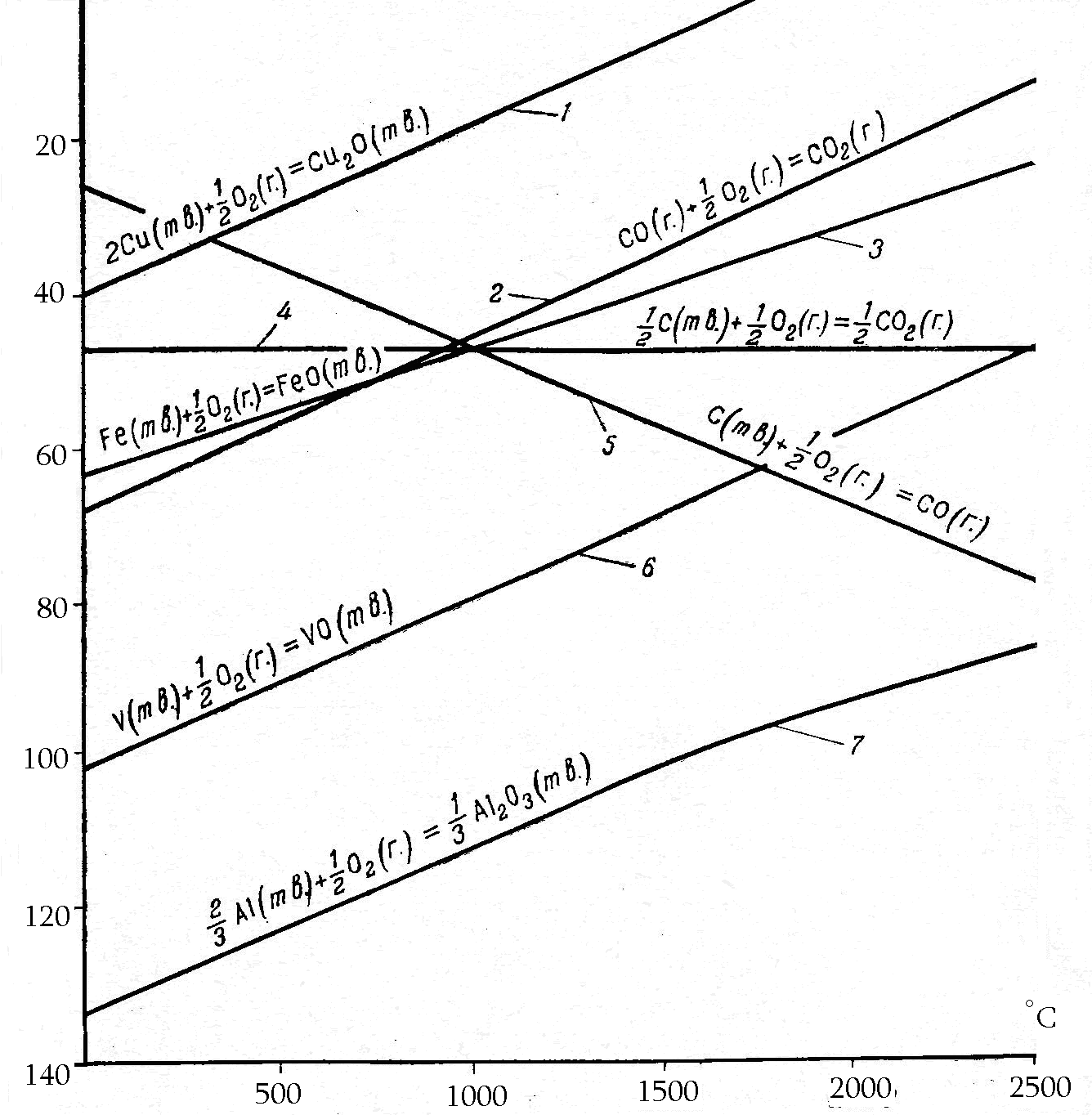
Рис.1. Изменение Go реакций образования оксидов при изменении температуры.
Важные выводы на основе диаграммы можно сделать о вероятности процессов, протекающих при выплавке чугунов и сталей. С точки зрения термодинамики процесс: «FeO» + СО = Fe + СО2 можно осуществить при температурах ниже 600оС, однако, при таких условиях не достигается энергия активации указанного процесса, а при Т > 600оС равновесие процесса будет смещаться влево, т.е. Fe будет окисляться за счёт СО2. «FeO» при Т > 900оС может быть восстановлен за счёт углерода. В соответствии с принципом наименьшей энергии и с учётом невозможности сосуществования в системе при Т > 600оС пары Fe + СО2 , процесс восстановления «FeO» описывается уравнением: «FeO» + С = Fe + СО, а рост температуры в системе способствует увеличению выхода продукта реакции.
Ещё одним распространённым восстановителем является молекулярный водород. Для реакции Н2(г.) + О2(г.) = Н2О(г.) ( S < 0) и, следовательно, с ростом температуры значения Go этого процесса увеличиваются (значение Go- уменьшается), т.е. изменения Go с температурой для этого процесса аналогичны графику Go – Т для взаимодействия СО(г.) + О2(г.) = СО2(г.) (рис. 1). Так как Go298 для обоих рассматриваемых процессов близки: -228,7 и - 257,4 кДж/моль, то можно утверждать, что Н2(г.), как восстановитель, будет более предпочтителен по сравнению с СО(г.),, если проведение процесса регламентируется температурами ниже 800оС. Это связано с тем, что при практически одинаковой термодинамике процессов с участием Н2(г.) и СО(г.), процессы с участием Н2(г.) будут иметь более низкую энергию активации. В связи с этим при Т < 700оС с помощью Н2(г.) может быть восстановлен не только Cu2O, но «FeO».
Выбор прекурсора для получения простого вещества не менее важен, чем выбор восстановителя. Так, например, процессы восстановления UO3 и UF6 с помощью Н2(г.) при с.у. имеют значение Go298 равные, соответственно, 467,4 и 421,6 кДж/моль, т.е. термодинамически практически невозможны. Повышение температуры в процессе восстановления UO3 вплоть до 2000оС не компенсирует различие в значениях энергии Гиббса прекурсоров и продуктов реакции, т. к. зависимости Go – Т реакций Н2(г.) + О2(г.) = Н2О(г.) и U + 1,5O2 = UO3 выражаются практически параллельными прямыми (как прямые 2 и 6 на рис 1). В то же время Go процесса: Н2(г.) + F2(г.) = 2НF(г.) практически не изменяется с ростом температуры (S реакции близко к нулю также как для реакции Ств. + О2(г.) = СО2(г.) - рис.1 прямая 4). В связи с тем, что для реакции U + 3F2(г.) = UF6(г.) ( Sо < 0), с ростом температуры в системе значения Go резко возрастают, процесс восстановления UF6(г.) водородом становится возможен уже при Т > 600оС.
В качестве восстановителей могут применяться и другие простые или сложные вещества, например, металлы. Это целесообразно только в том случае, когда, либо Go298 восстанавливаемых форм имеет очень низкое значение и, следовательно, при использовании ранее рассмотренных восстановителей, невозможно добиться отрицательного значения Go реакции синтеза простого вещества в пределах допустимого варьирования параметров состояния системы, либо за счёт протекания последовательных или параллельных процессов продукт реакции содержит недопустимую концентрацию примесей (например, углерод, растворённый в целевых продуктах или карбиды различных элементов). Возможность осуществления процессов рассматриваемого типа, на практике, также как в предыдущих случаях регламентируются термодинамической возможностью протекания взаимодействия при выбранных параметрах состояния систем и скоростью реакций, протекающих в этих системах. С учётом кинетических факторов в качестве восстановителей часто применяются металлы с низкой энергией кристаллической решётки (натрий, магний, алюминий). В свою очередь выбор восстановителя диктует выбор восстанавливаемой формы. Если же задана восстанавливаемая форма, то тогда она диктует выбор восстановителя. Поясним последние тезисы на примерах:
а) Какие формы целесообразно восстанавливать с помощью натрия?
Go298 образования Na2O и NaCl равны, соответственно – 379,3 и -384,
Тогда при с.у. для процессов:
Al2O3 + 6Na = 2Al + 3Na2O (Go298 = - 3379,3 + 1583,3 = 445,4 кДж)
Al2Cl6 + 6Na = 2Al + 6NaCl (Go298 = - 6384,3 + 1258 = - 1047,8 кДж)
Из представленных данных видно, что при с.у. восстановить оксид алюминия с помощью натрия невозможно. Повышение же температуры в этой системе приведёт к переходу натрия в газообразное состояние и, следовательно, к дальнейшему смещению равновесия рассматриваемого процесса в сторону исходных веществ. В то же время хлорид алюминия (с точки зрения термодинамики) может быть восстановлен с помощью натрия уже при с.у., а с учётом низкой энергии активации этого процесса, указанная реакция с высокой скоростью протекает при Т > 150oC. Аналогично можно показать, что с помощью натрия выделить ниобий или титан из их оксидов невозможно, но это можно сделать, если в качестве прекурсора использовать комплексный фторид или галогениды:
K2[NbF7] + 5Na = 2KF + 5NaF + Nb (Go298 << 0)
TiCl4 + 4Na = 4NaCl + Ti (Go298 << 0)
Очевидно, что преимущество галогенидных фаз перед оксидными (с термодинамической точки зрения) для рассматриваемых процессов заключается в их меньшей термодинамической стабильности (сравните Go298 Al2O3 и Al2Cl6) и в большем числе молей продуктов реакции (при равном числе молей образующегося металла число молей образующегося оксида натрия в два меньше, чем число молей галогенида). Все это, с учётом необходимости проведения указанных процессов при температурах ниже температуры кипения восстановителя, свидетельствует, что щелочные металлы при синтезе простых веществ могут быть использованы преимущественно в реакциях, где в роли окислителя выступает галогенидная фаза.
б) Какие формы целесообразно восстанавливать с помощью алюминия?
Go298 образования Al2O3 и Al2Cl6 равны, соответственно -1583,3 и -1258 кДж/моль, а Go298 образования VCl4 и VO2 составляют, соответственно -506 и -658,5 кДж/моль. Рассмотрим термодинамику восстановления соединений ванадия с помощью Al:
3VO2 + 4Al = 2Al2O3 + 3V (Go298 = -21583,3 + 3658,5 = -1191,7 кДж)
3VCl4 + 4Al = 2Al2Cl6 + V (Go298 = -21258,1 + 3506,4 = -997,0 кДж), т.е. при стандартных условиях возможно протекание обоих процессов и при их инициировании (для достижения энергии активации) они протекают с очень высокой скоростью. Однако получение переходных металлов методом алюмотермии имеет один существенный недостаток: алюминий со многими d-элементами образует устойчивые интерметаллиды, состав и структура которых будет рассмотрена в разделе 2. Их удаление из системы – длительный и трудоёмкий процесс, поэтому использование алюминия в качестве восстановителя соединений переходных элементов оправдано только в том случае, когда использование других восстановителей малоэффективно.
в) Одним из способов, обеспечивающих самопроизвольное течение процессов получения простых веществ, является их целенаправленное конструирование, заключающиеся в том, что помимо искомого простого вещества планируется формирование и другие фазы, характеризующиеся низким значением Go образования, что и обеспечивает отрицательное значение Go суммарного процесса, например:
Ca3(PO4)2 + 5C + 3SiO2 = P2 + 3CaSiO3 + 5CO - в данном случае, помимо низкого значения Go образования CaSiO3, смещению равновесия вправо способствует значительное повышение энтропии системы при переходе от прекурсоров к продуктам реакции. Очевидно, что рост температуры в этой системе будет ещё в большей степени смещать равновесие в указанном направлении и, следовательно, высокая энергия активации рассматриваемого процесса не будет препятствовать получению продуктов реакции: он будет протекать при Т > 1000оС, практически, со 100% выходом.
Аналогичные приёмы используются и при получении щелочных и щелочноземельных металлов:
4KCl + Si + 3CaO = 2CaCl2 + CaSiO3 + 4K
6RbCl + 2Al + 4CaO = 3CaCl2 + CaAl2O4 + 6Rb
6CaO + 2Al = Ca3Al2O6 + 3Ca - т.е. протеканию реакций в прямом направлении при высоких температурах способствуют высокая термодинамическая стабильность сложных веществ, которые являются продуктами реакции, и значительный рост энтропии в процессе формирования продуктов взаимодействия, за счёт образования паров простых веществ.
К пирометоды получения простых веществ также могут быть основаны на реакциях разложения бинарных веществ:
B2H6 = 2B + 3H2 ; CH4 = C + 2H2 ; SiI4 = Si + I2 ;
сложных веществ: NH4NO2 = N2 + 2H2O ; (NH4)2[PtCl6] = Pt + 2NH4Cl + 2Cl2 или в процессе, который включает стадию окисления прекурсора и разложение продуктов реакции: HgS + 3/2O2 = HgO + SO2, - в момент образования оксид ртути разлагается за счёт теплоты, выделяющейся на первом этапе процесса: HgO = Hg + O2.
На росте энтропийного фактора могут быть основаны и способы получения простых веществ, использующие в качестве и восстановителя и окислителя соединения элемента, простое вещество которого синтезируется. Очевидно, что в этом случае успех будет зависеть от того, на сколько низка стабильность прекурсоров по сравнению с побочным продуктом реакции. Например:
2Cu2O + Cu2S = 5Cu + SO2 (Go298 = 87,1 кДж)
2PbO + PbS = 3Pb + SO2 (Go298 = 234,0 кДж) – как видно из представленных данных, при стандартных условиях самопроизвольное протекание указанных процессов в прямом направлении невозможно. Однако, в связи с положительным знаком S прямого процесса, Go обеих реакций становится меньше нуля уже при Т > 600оС, а удаление SO2 из системы будет способствовать дальнейшему смещению равновесия в сторону продуктов реакции.
В основе ещё одного из методов лежат изменения стабильности подобных форм соединений элементов одной подгруппы, например:
H2SO3 + H2SeO3 = Se + H2SO4 + H2O
H2SO3 + H2SeO3 = S + H2SeO4 + H2O - так как прекурсоры обоих процессов одинаковы, вероятность протекания процесса в водном растворе будет предопределяться относительной стабильностью ионов SO42- и SeO42-. В соответствии с изменениями радиусов центральных ионов в этих частицах, можно сделать вывод, что SO42- долее стабильна по сравнению с SeO42- и, следовательно, из двух параллельных процессов в системе будет протекать, преимущественно, первый, что позволит получать этим способом селен.
Простые вещества практически всех элементов (за исключением инертных и благородных газов) могут быть получены с использованием различных вариантов пирометодов. Общим недостатком этих способов является низкая чистота получаемого простого вещества, за счёт протекания в системах трудно контролируемых параллельных и последовательных процессов. Поэтому, получаемые указанными методами, простые вещества носят техническое название «черновые», смысл которого заключается в том, что образующийся продукт реакции содержит переменную долю примесей и для получения искомого вещества «черновой» продукт необходимо подвергнуть дополнительной обработке.
В промышленных условиях выбор прекурсоров ограничен распространёнными рудами элемента, простое вещество которого необходимо получить. Так как многие d- и f-элементы относятся к редким и рассеянным, в качестве исходного сырья для получения их простых веществ и соединений применяются руды с содержанием элементов 0,5 – 2 моль.%. В этом случае первый этап процесса – выделение первичного концентрата соединений определённого элемента – проводится с помощью гидрометодов. При этом, в ряде случаев, полученные концентраты можно использовать для получения простых веществ непосредственно в водных растворах, например, используя методы цементации (восстановление ионов в растворах, с использованием в качестве восстановителей различные металлы):
CuSO4(p-p.) + Fe = Cu + FeSO4(p-p.)
Na[Au(CN)2](p-p.) + Zn = Au + Na2[Zn(CN)4](p-p.)
Химические методы, основанные на процессах окисления прекурсоров, используются, преимущественно, при получении простых веществ галогенов и протекают при с.у.:
MnO2 + 4HCl = MnCl2 + 2H2O
2KI + Cl2 = 2KCl + I2
Электрохимические методы получения простых веществ в принципе позволяют получить простые вещества многих элементов, однако их практическое использование, зачастую, ограничено более высокой себестоимостью по сравнению с химическими способами получения данного простого вещества (даже с учётом очистки «черновой» продукции). Поэтому указанные методы применяются только тогда, когда это экономически оправдано, т.е. отсутствуют более дешёвые химические способы искомой фазы. В частности, электролизом расплавов различных фаз или многокомпонентных расплавов получают алюминий, бериллий, литий, натрий, кальций и другие металлы, имеющие низкие значения стандартных электродных потенциалов, а также F2. Электролизом растворов получают металлы, расположенные в ряду напряжений правее марганца, а также галогены (за исключением F2). Разработаны электрохимические методы синтеза простых веществ некоторых халькогенов и пиктогенов.
1.2. Состав и строение простых веществ.
Состав и строение простых веществ предопределяется электронной структурой образующих их атомов и параметрами состояния системы. Как отмечалось выше, конечной формой всех простых вещества является совокупность атомов. Температурный интервал существования такой формы для конкретного элемента будет предопределяться стабильностью других форм, образующихся в результате ассоциации атомов. В свою очередь стабильность молекулярных, полимерных, кластерных или кристаллических форм простых веществ данного элемента предопределяется электронным строением его атомов и энергией электронов на их валентном уровне.
Так для элементов главной подгруппы VIII группы газообразная атомная фаза существует в широком температурном интервале при стандартном давлении. Для серы атомный пар образуется только выше 1500оС, а диссоциация молекул N2 наблюдается только выше 3000оС.
При охлаждении газообразных атомных форм возможно протекание процессов атомной ассоциации различных типов:
1. Межатомное взаимодействие дисперсионного типа, имеющее небольшую энергию и приводящее к образованию ассоциатов при низких температурах. Жидкая фаза таких веществ состоит из атомных кластеров, из которых при температуре кристаллизации формируются плотноупакованные кристаллические фазы (структура ГЦК или ГПУ, рис 2а). Такие формы простых веществ при различных температурах и давлениях обнаружены у элементов главной подгруппы VIII группы.
2. Образование различных по количественному составу молекулярных форм. Образование таких форм при различных температурах установлено для всех элементов, за исключением элементов главной подгруппы VIII группы, а также бериллия и магния. Температурный интервал стабильности таких форм зависит от энергии связи в молекуле и относительной устойчивости других аллотропных или полиморфных модификаций по сравнению с данной молекулярной формой. Эксперимент показывает, что первым этапом межатомного взаимодействия является образование двухатомных молекул. Они появляются в системе при тем больших температурах, чем выше энергия связи в молекуле, т.е. чем ниже значение изменения энтальпии при образовании связи в молекуле. При охлаждении молекулярного пара возможны несколько типов дальнейшего организации вещества.
1. Если энергия межатомного взаимодействия в молекуле превышает энергию межмолекулярного взаимодействия (т.е. отсутствует возможность полимеризации молекул с частичным разрывом в них химических связей), то состоящее из таких молекул газообразное простое вещество при охлаждении будет конденсироваться, за счёт межмолекулярного взаимодействия дисперсионного типа. Результатом этого будет образование молекулярных кластеров (жидкая фаза) и молекулярных кристаллов. Примерами таких веществ являются H2, O2, N2, Cl2 и т.д. (рис. 2б).
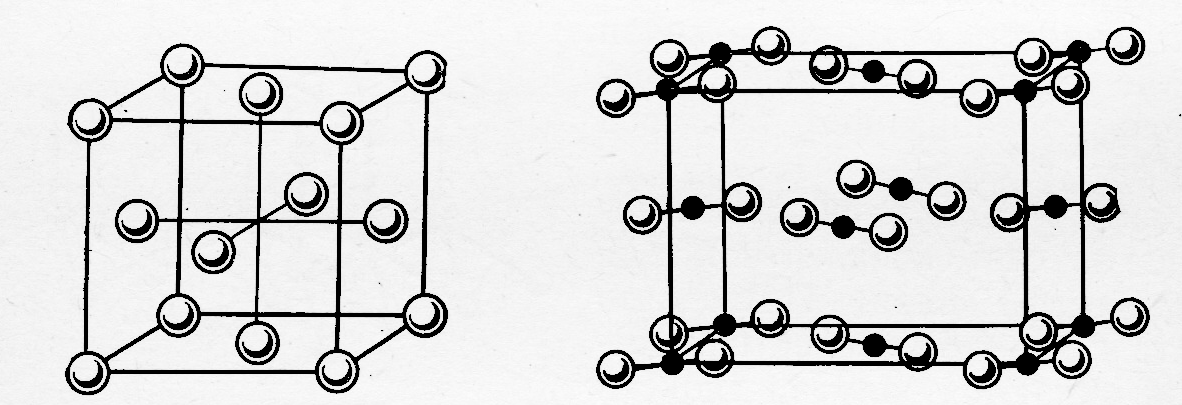
а) б)
Рис.2. а) – кристаллическая структура аргона, б) – кристаллическая структура йода.
2. Если же энергия межмолекулярного взаимодействия выше, чем энергия связи между атомами в молекуле, то происходит образование более крупных частиц (молекул или кластеров с ковалентным характером связи между атомами).
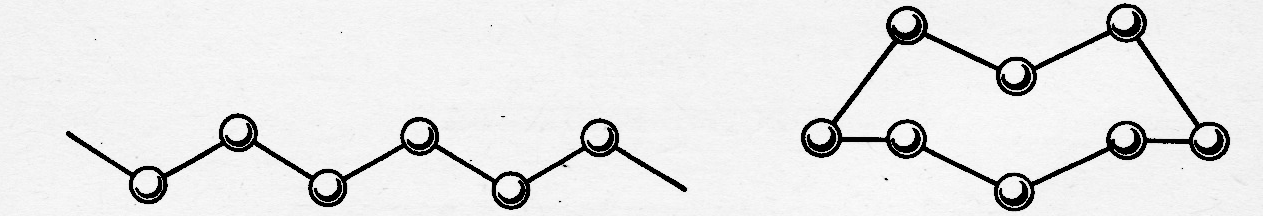
а) б)
в)
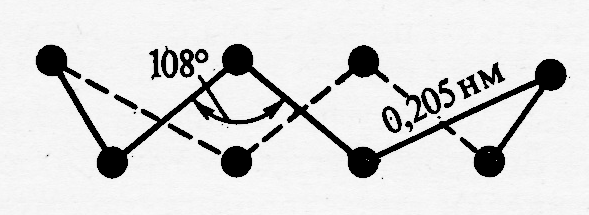
Рис. Структура модификаций серы: а) – пластическая (полимерная сера, б) и в) – различные способы изображения молекулы S8
Например, по мере снижения температуры атомный пар серы претерпевает следующие изменения: S S2 S4 S6 S8 Sn (ж.) S8(т.) – где Sn (ж.) – полимерная форма серы (рис.3), термодинамически стабильная в жидком состоянии при Т > 160оС, подчёркнуты формы, характерные для газообразного состояния этого вещества.
Аналогичные изменения наблюдаются и при охлаждении паров фосфора:
Р Р2 Р4 Рn(красный) Рn(чёрный) – подчёркнуты формы, характерные для газообразного состояния этого вещества.
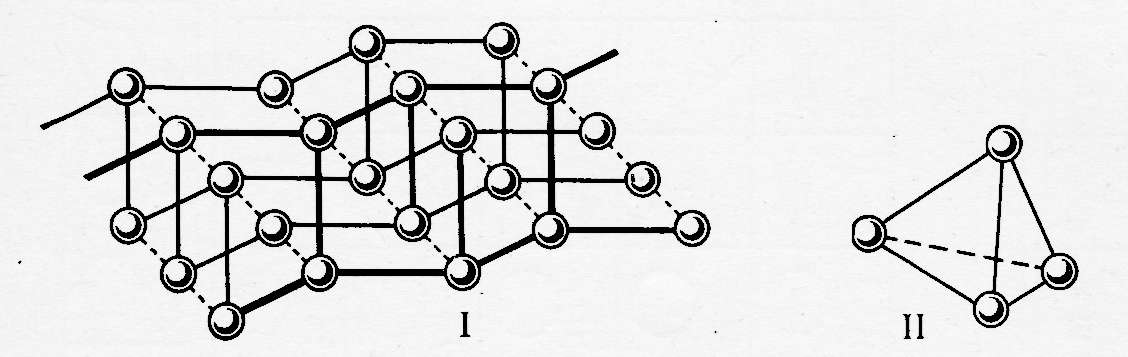
Рис. 4. Полимерная ( I - «чёрный фосфор») и молекулярная ( II - «белый фосфор») модификации фосфора
Среди полимерных форм простых веществ выделяют линейные (Sn), двумерные (Сграфит – рис.5) и трёхмерные (Рn(чёрный)) (рис.4).
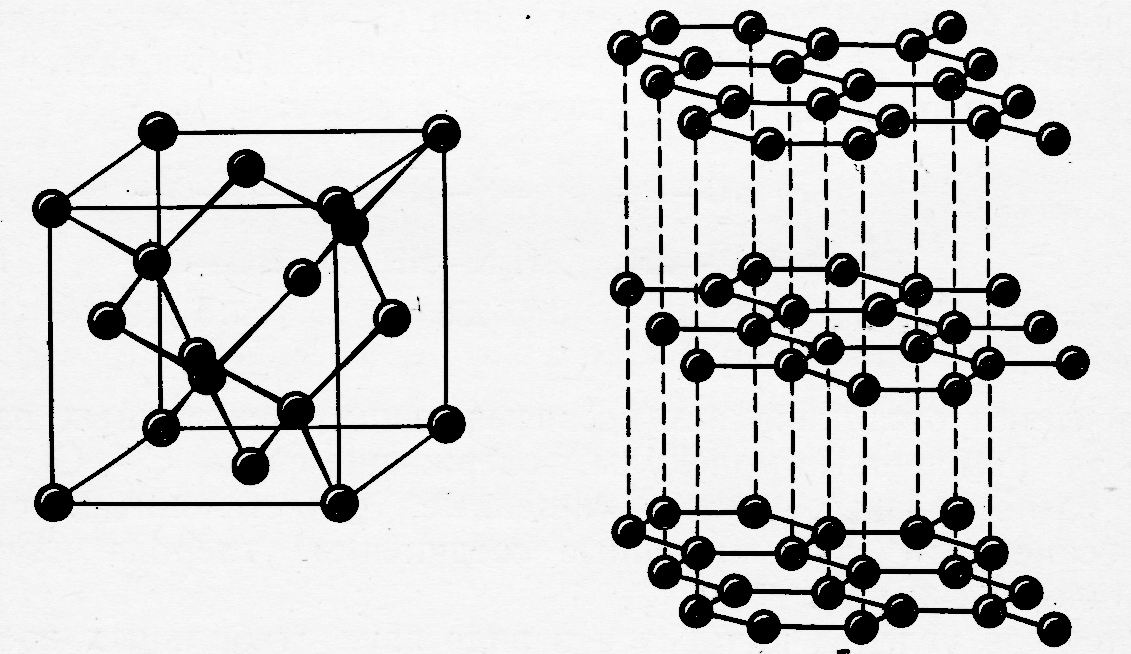
а) б)
Рис.5. Кристаллическая структура а) - алмаза, б) – графита.
Образование атомных кластеров с ковалентным локализованным или делокализованным («металлическим») характером связи. Из кластеров первого типа при температуре кристаллизации формируются кристаллы, атомы в которых соединены ковалентными, неполярными, двухцентровыми связями, направленными в пространстве в соответствии с типом гибридизации орбиталей взаимодействующих атомов. Для таких кристаллов характерны относительно низкие координационные числа (от 3 до 6). Классические вещества этого типа – диэлектрики, например алмаз (рис.5а). Однако с уменьшением потенциала ионизации атомов, вещества, сохраняя структуру с низкими значениями к.ч., приобретают полупроводниковые свойства, за счёт частичной делокализации электронной плотности химической связи (кремний, германий, бор и т.д.).
При делокализации химической связи, к.ч. в кластерах и кристаллах резко увеличивается до 8 – 12, что обеспечивает формирование трёх основных типов кристаллических решёток, характерных для металлов: объёмноцентрированной кубической (ОЦК) – к.ч.= 8, и двух плотноупакованных с к.ч. = 12 – гранецентрированной кубической (ГЦК) и с гексагональной плотнейшей упаковкой (ГПУ) (рис.6).
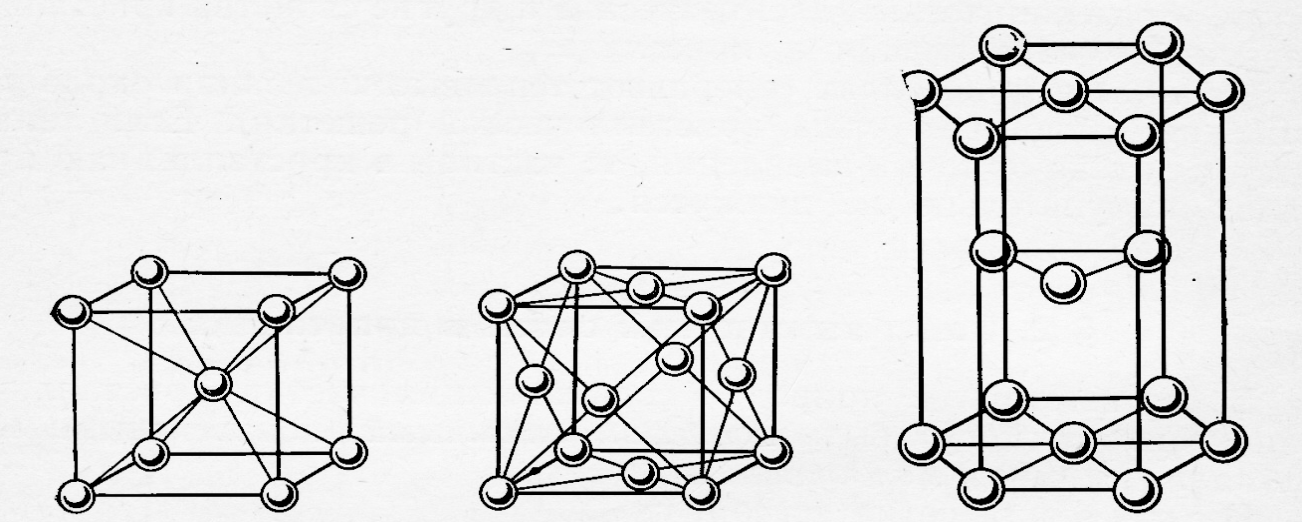
а) б) в)
Рис.6. Кристаллическая структура металлов: а) – ОЦК, б) – ГЦК, в) - ГПУ
Необходимо отметить, что существует целый ряд простых веществ в конденсированных состояниях с промежуточным типом взаимодействия между атомами. Например, галлий в жидком и газообразном состоянии – молекулярное вещество. При кристаллизации жидкой фазы в этом случае формируется псевдомолекулярная структура, в которой один из атомов Ga расположен на более коротком расстоянии от центрального атома, по сравнению с остальными. Структуры его полиморфных модификаций индивидуальны, хотя по электрофизическим свойствам они могут быть отнесены к металлам. Структура же висмута аналогична структуре чёрного фосфора, однако для этого простого вещества характерны свойства, характеризующие его как металл. С другой стороны многие переходные металлы имеют аномально высокую твёрдость, низкую пластичность, хрупкость, их температуры плавления достигают температур выше 3000оС и т.д., т.е. целый ряд свойств, которые не могут быть объяснены в рамках концепции «металлической» связи. Все указанные примеры показывают, что единственным критерием стабильность тех или иных форм является минимум энергии системы при заданных параметрах её состояния. Этот минимум достигается всеми возможными ресурсами, которые имеются у атомов, формирующих ту или иную форму. Например, в кристаллах простых веществ переходных элементов только часть связей, образованных за счёт перекрывания наибольших по энергии орбиталей делокализована, тогда как при перекрывании d-орбиталей образуются локализованные двухцентровые связи, направленные в пространстве которых предопределяется типом их гибридизации.
2. Бинарные соединения.
Нахождение в одной системе атомов двух различных элементов расширяет число возможных путей достижения в ней минимума энергии. Это связано с тем, что атомы каждого элемента индивидуальны и в пределах атомов одного вида могут быть образовано только небольшое количество форм простых веществ, которые были рассмотрены выше. По мере накопления различий между атомами число таких форм будет увеличиваться, так как атомы будут устранять «недостатки» друг друга, связанные с их фиксированным строением. Например, атомы элементов главных подгрупп I и II групп имеют большое число валентных орбиталей, но малое количество валентных электронов. Это приводит к тому, что при образовании простых веществ локальный минимум энергии в таких системах при с.у. достигается только при обобществлении валентных электронов – суммарная энергия такой связи не велика и образующиеся формы химически не стабильны. С другой стороны низкая стабильность молекулы F2 объясняется избытком электронов в системе (число электронов значительно превышает число валентных орбиталей). Очевидно, что при наличии в одной системе атомов двух указанных видов между ними будет происходить взаимодействие, при котором каждый из них свой «недостаток» превратит в «достоинство», в результате чего энергия бинарной системы окажется значительно ниже, чем суммарная энергия смеси двух простых веществ. Как уже отмечалось, указанные изменения в системах будут происходить тем в большей степени, чем значительнее различия в строении взаимодействующих атомов. Рассмотрим последовательный ряд бинарных фаз, по мере роста различий в строении образующих их атомов.
2.1. Твёрдые растворы металлического типа
Характерной особенностью твёрдых растворов с делокализованным характером связи является то, что качественные и количественные характеристики данных фаз изменяются аддитивно при изменении в них соотношения компонентов. Критериями образования непрерывного ряда твёрдых растворов является близость: а) электронного строения атомов элементов, образующих рассматриваемую фазу; б) кристаллохимического строения исходных простых веществ; в) радиусов атомов, входящих в состав бинарной фазы и их электроотрицательностей. В соответствии с правилом Руайе, радиусы атомов, входящих в состав неограниченного твёрдого раствора не должны отличаться более чем на 15%, что, с учётом подобия остальных перечисленных выше факторов, предопределяет одинаковый тип кристаллических решёток прекурсоров. Если различия в значениях радиусов больше 15% то, в таких системах наблюдается образование ограниченных твёрдых растворов (таблица 1).
Таблица 1. Состав твёрдых растворов на основе -титана
|
элемент |
V |
Cr |
Mn |
Fe |
Co |
Ni |
|
Растворимость в -Ti ат.% |
100 |
100 |
28 |
20 |
13 |
10 |
В связи с близкими значениями орбитальных атомных радиусов и сходной электронной оболочке валентного уровня, непрерывные твёрдые растворы образуют между собой простые вещества, элементы которых входят в одну и ту же побочную подгруппу: Ti Zr Hf , V Nb Ta, Cr Mo W, а также соседних подгрупп. В то же время в главных подгруппах d- и f-сжатие в значительно меньшей степени влияет на изменение радиуса атома и он быстро возрастает в подгруппе сверху вниз. Это приводит к тому, что, несмотря на аналогичность строения электронных оболочек атомов и кристаллов простых веществ, не всегда удаётся получить твёрдые растворы, в состав которых входят элементы одной и той же главной подгруппы. Например, в первой главной подгруппе наиболее близки атомные радиусы у K, Rb и Cs, которые между собой образуют непрерывные ряды твёрдых растворов. Различие атомных радиусов натрия и калия порядка 30% и они между собой образуют ограниченные твёрдые растворы и интерметаллические соединения. Разница в атомных радиусах натрия с одной стороны, и рубидия и цезия – с другой больше, чем в случае пары K – Na. Это приводят к тому, что при взаимодействии Na с Rb или Cs образование твёрдых растворов не наблюдается, а формируются интерметаллиды различного состава. Наиболее интересно поведение лития по отношению к другим щелочным металлам: в этих бинарных системах не наблюдается образование не только твёрдых растворов, но и интерметаллидов, а в жидком состоянии в этих системах происходит расслоение.
С термодинамической точки зрения образование металлических твёрдых растворов характеризуется очень небольшим изменением энтальпии, при этом, зачастую, Н > 0. Следовательно, главную роль в формировании этих твёрдых растворов играет энтропийный фактор, тогда рост температуры системы будет способствовать расширению областей ограниченных твёрдых растворов, которые выше температуры ликвидуса могут быть неограниченно растворимы друг в друге.
2.2. Интерметаллические соединения.
Основное отличие процесса образования интерметаллических соединений от твёрдых растворов заключается в том, что в его результате возникает новый индивид, характеризующийся отличной от исходных фаз структурой и свойствами. По мере нарастания различий в характеристиках атомов исходных элементов и простых веществ характер взаимодействия в системах усложняется, и возникают фазы различного типа, на примере которых удобно рассматривать эволюцию веществ по мере роста различий у атомов – партнёров. Эта последовательность условно может быть представлена в виде ряда: соединения Курнакова фазы Лавеса электронные соединения Юм-Розери фазы внедрения. Рассмотрим более подробно строение и причины образования указанных форм.
2.2.1. Соединения Курнакова
В 1914 году Курнаков, исследуя систему Cu – Au, установил, что при медленном охлаждении, первоначально сформировавшийся в ней твёрдый раствор разрушается с образованием соединений, средний состав которых Cu3Au и CuAu. В свою очередь эти фазы образовывали твёрдые растворы с избытком одного из компонентов. Строение этих фаз было установлено позднее методом РФА (рис.7).
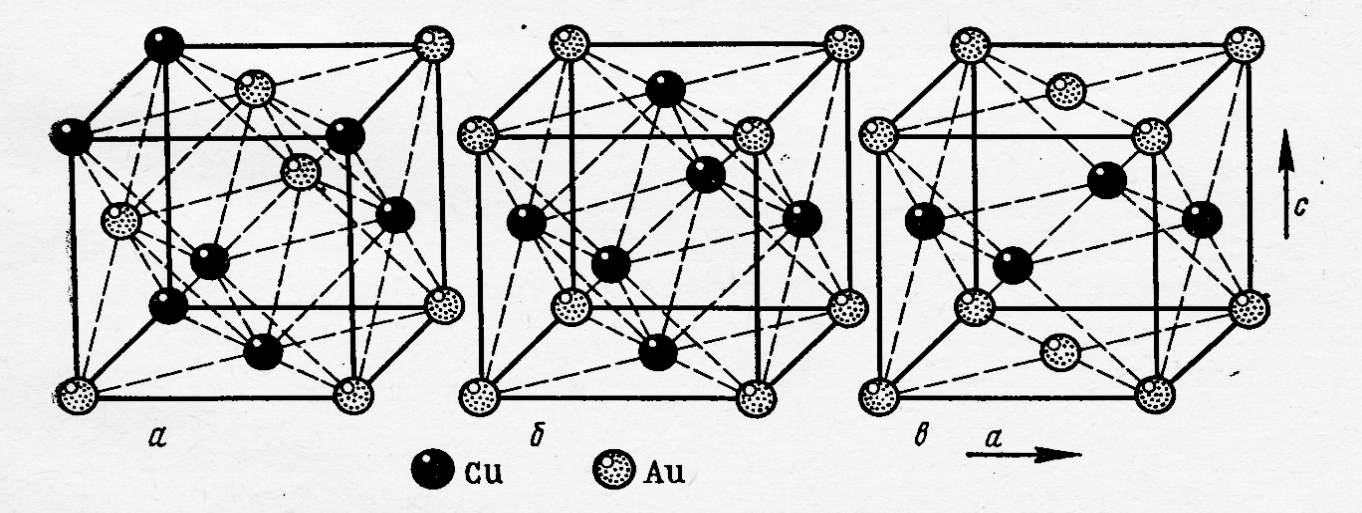
Рис.7. Строение элементарных ячеек: а) твёрдый раствор системы Cu – Au; б) фазы Cu3Au; в) фазы CuAu
Исходные металлы имеют структуру ГЦК и в жидкой фазе имеют неограниченную растворимость. Неограниченные твёрдые растворы этой системы можно получить методом закалки от температуры кристаллизации. В этих фазах атомы меди и золота статистически неупорядочено занимают узлы кристаллической решётки. Если же расплав кристаллизуется в равновесных условиях или полученный метастабильный твёрдый раствор подвергается отжигу, то происходит упорядочение атомов в кристаллической решетке, результат которого показан на рисунках 7б и 7в. Эти типы упорядочения соответствуют определённым количественным составам фаз, строение которых отлично от исходного твёрдого раствора. В фазе изображённой на рисунке 7б, атомы Au находятся в вершинах куба, и каждый из них принадлежит элементарной ячейке на 1/8, тогда в ячейке число атомов Au составляет 1/88 = 1. В то же время атомы меди занимают центры граней, т.е. принадлежат элементарной ячейке на , тогда число этих атомов в элементарной ячейке 6 = Следовательно, состав первой из фаз Cu3Au.
Во второй фазе атомы Au занимают вершины куба и центры двух противоположных граней, а атомы меди располагаются в центрах четырёх граней. Тогда число атомов Au в элементарной ячейке: 1/88 + 2 = 2, а атомов Cu: 4 = 2, следовательно, состав второй фазы – CuAu. В отличии от Cu3Au, которая имеет кубическую структуру, фаза CuAu имеет тетрагональную симметрию элементарной ячейки. Это связано с тем, что радиус атома меди меньше радиуса атома золота и, следовательно, отношение с/а у этой фазы меньше единицы (с/а = 0,932). Так как формирование указанных фаз происходит при относительно низких температурах, степень их упорядоченности в реальных условиях не более 90%. В отличие от процесса образования твёрдого раствора, образование фаз Курнакова сопровождается снижением, как энтальпии, так и энтропии системы.
В настоящие время установлено, что образование соединений Курнакова характерно для многих элементов Периодической системы, при этом простые вещества, выступающие в качестве прекурсоров, могут относиться, как к металлам, так и к неметаллам: AuAg3, AuAg, MgAg3, Fe3Al, VNi3, PtCu, VCo3, MnAu3, Ni3Au, Ti3Al, (Me2)3O, Me3O (где Ме = Ti, V, Cr и т.д.). Соединения Курнакова качественно сохраняют тип кристаллической структуры минимум одной из исходных фаз, однако свойства продуктов реакций значительно отличаются от свойств исходных компонентов. Например, в отличие от твёрдых растворов материалы на основе этих фаз характеризуются повышенной хрупкостью. Это их свойство используют для получения порошков сплавов различного состава. Фазы Курнакова, в состав которых входят атомы железа обладают высокой магнитной проницаемостью (FeNi3, Fe3Al, FeCo), а содержащие атомы ниобия являются высокотемпературными сверхпроводниками (Nb3Sn, Nb3Al, Nb3Ga) и т.д..
Как следует из представленных примеров, различия атомов в электроотрицательности в данном случае не играет решающей роли (AuAg3 MgAg3). Анализ показывает, что основным фактором, приводящим к упорядочению в этих системах, является размерный.
2.2.2. Фазы Лавеса
Общими признаками, отличающие фазы Лавеса от других бинарных интерметаллидов, является: а) определённый состав, отвечающий формуле АВ2; б) плотноупакованная структура с к.ч. 12; в) размерный критерий стабильности. Следует отметить, что роль размерного фактора в стабильности фаз Курнакова и фаз Лавеса различен: если условием образования соединений Курнакова является относительная близость атомных радиусов, то для фаз Лавеса – их существенное различие (в идеальном случае объём атома А в два раза больше объёма атома В или rA: rB 1,26). При таком или близком соотношении радиусов атомы А занимают в кристаллической решётке фазы два места плотнейшей упаковки, и атомы В – одно (рис.8.), что отвечает формальному составу АВ2. Например, соединение MgCu2 (рис.8.): rMg = 0,160 нм, rCu = 0,128 нм, тогда VMg : VCu = 1,95, что отвечает условиям формирования фаз рассматриваемого типа.
Как видно из рисунка 8., атомы магния образуют плотноупакованную структуру типа ГЦК. Ещё четыре атома магния занимают центры четырёх из восьми октанов. Тетраэдры из атомов меди (Cu4) занимают четыре из оставшихся октанов, в которых они расположены таким образом, что центры тетраэдров совпадают с центрами этих октанов.
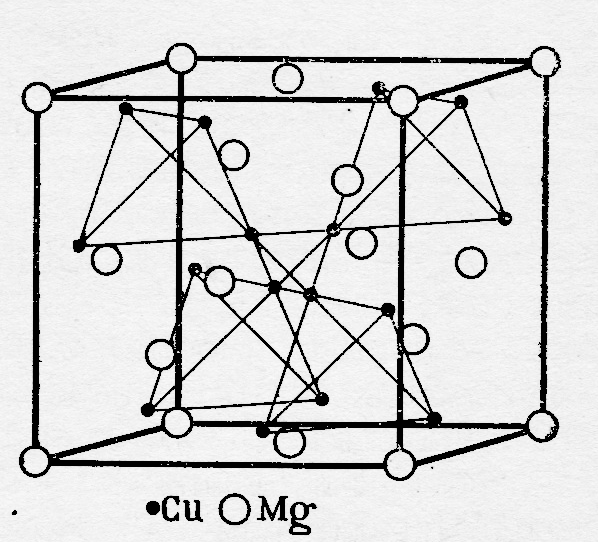
Рис.8. Элементарная ячейка MgCu2
Таким образом в фазах Лавеса сосуществуют связи А – В, А – А и В – В, т.е. рассматриваемые соединения относятся к гетеродейсмичным (содержащими связи нескольких типов).
Таким образом, фазы Лавеса, по сравнению с соединениями Курнакова, являются следующей ступенью организации вещества – несмотря на то, что критерием их образования остаётся размерный фактор, они отличаются от прекурсоров не только по физическим и физико-химическим свойствам, но и по типу структуры.
2.2. Соединения Юм – Розери.
Ряд интерметаллических соединений разнообразного качественного и количественного состава формируются на основе фактора формальной электронной концентрации (ФЭК). ФЭК определяется, как отношение общего числа валентных электронов (которое в данном случае принимается равным номеру группы) к числу атомов в формульной единице. Фазы указанного типа называются электронными соединениями Юм – Розери. Их образование фиксируется в системах, содержащих, с одной стороны элементы побочных подгрупп I и VIII групп, и с другой – элементы подгрупп: IIВ, IIIА и IVА. Состав рассматриваемых фаз определяется только значением ФЭК, которое для термодинамически стабильных форм может принимать только несколько значений, в частности:
1) ФЭК = 21/14 = 3/2 – структура типа ОЦК (-форма);
2) ФЭК = 21/13 – кубическая структура с 52 атомами в элементарной ячейке ( – форма);
3) ФЭК = 21/12 = 7/4 – структура типа ГПУ (-форма).
Соединения Юм – Розери, имеющие один и тот же структурный тип, могут иметь различный состав (таб.2).
Таблица 2. Состав и структура соединений Юм – Розери.
|
состав |
n-число электронов |
nа-число атомов |
ФЭК в n/ nа |
тип структуры |
|
CuZn |
1 + 2 |
2 |
3/2 |
- форма |
|
Cu3Al |
3 + 3 |
4 |
3/2 |
- форма |
|
Cu5Zn8 |
5 + 16 |
13 |
21/13 |
– форма |
|
Cu9Al4 |
9 + 12 |
13 |
21/13 |
– форма |
|
Cu31Sn8 |
31 + 32 |
39 |
21/13 |
– форма |
|
*Co5Zn21 |
0 + 42 |
26 |
21/13 |
– форма |
|
AgCd3 |
1 + 6 |
4 |
7/4 |
- форма |
|
Ag5Al3 |
5 + 9 |
8 |
7/4 |
- форма |
|
Cu3Sn |
3 + 4 |
4 |
7/4 |
- форма |
*в соединениях, содержащих элементы VIII группы, число валентных электронов у последних принимается равным нулю. Во всех случаев учитывается число электронов, расположенных у атомов на s- и p-подуровнях.
Смысл влияния ФЭК на состав и структуру соединений данного типа можно понять на основе зонной теории химической связи. Согласно этой теории каждой кристаллической структуре отвечает совокупность молекулярных орбиталей, которые можно разбить на отдельные группы (зоны) в соответствии с их энергией. В кристаллах с делокализованной связью валентные атомные орбитали прекурсоров формируют единую зону молекулярных орбиталей, в которой валентная зона и зона проводимости энергетически не разделены. В соответствии с принципом наименьшей энергии наиболее стабильному состоянию такой системы будет отвечать состояния с наполовину заполненной энергетической зоной (тогда, формально, все валентные электроны располагаются на связывающих орбиталях). При большем числе электронов в системе её стабильность снижается и при достижении определённого максимума формирование данной структуры становится невозможным. Рассмотрим последовательность формирования фаз в системе Cu – Zn. У простого вещества элемента Cu структура ГЦК с наполовину заполненной s-зоной. Введение в систему Zn, у которого на валентной s-орбитали электронов в два раза больше, чем у атома меди, приведёт к появлению электронов на, формально, разрыхляющих молекулярных орбиталей s-зоны, что повысит энтальпию системы. Следовательно, самопроизвольное образование твёрдого раствора в данном случае будет регламентироваться энтропийным фактором, возможности которого не беспредельны. В связи с этим растворимость цинка в меди ограничена, и отвечает области ФЭК от 1 до 1,4. Стабилизация системы при дальнейшем увеличении в ней концентрации цинка невозможна без изменения структуры зоны, а следовательно, и структуры элементарной ячейки фазы. В кристаллах с ОЦК структурой наблюдается формирование непрерывной sp-зоны, с участием s-орбиталей и одной из трёх p-орбиталей, расположенных в одной плоскости. Тогда нижняя по энергии sp-зона при общем числе атомов в системе 2n будет иметь 3n орбиталей, а её заполнению на будет отвечать наличие у 2n атомов 3n. Это условие в рассматриваемой системе отвечает составу CuZn (табл.2), имеющей ФЭК = 1,5 /атом, т.е. - форма. Область её гомогенности лежит в пределах ФЭК от 1,42 до 1,56 /атом.
Дальнейшее увеличение концентрации цинка в системе должно привести к тому, что только его p-орбитали будут принимать участие в формировании sp-зоны по описанному выше способу. Тогда, с математической точки зрения, в формировании sp-зоны на следующем этапе должны принимать участие s-орбитали 5n атомов меди, а также по одной s- и p-орбитали от каждого из 8n атомов цинка. Тогда общее число орбиталей в sp-зоне: (5 + 8 + 8 = 21) n и число электронов тоже 21n, т.е. sp-зона при составе Cu5Zn8 будет заполнена на половину ( – форма, область гомогенности по ФЭК = 1,60 – 1,65 /атом). Не трудно показать, что следующее удачное с точки зрения сочетание числа орбиталей в sp-зоне и числа валентных электронов, будет отвечать составу CuZn3, в чём мы рекомендуем убедиться читателям. ФЭК для этой фазы составит 1,75 /атом ( – форма с областью гомогенности по ФЭК от 1,75 до 1,89 /атом).
2.2.4. Фазы внедрения.
К фазам, образование которых также в значительной степени определяется размерами атомов, относятся и фазы внедрения электроотрицательных атомов в кристаллическую структуру металлов. Фазы этого типа образуют только d- и f-элементы с не завершённым d или f-подуровнем. Рассмотрим этапы процесса взаимодействия такого металла с неметаллом. На первом этапе, при низкой концентрации атомов электроотрицательного элемента в системе происходит формирование неупорядоченного твёрдого раствора, этот процесс имеет Н > 0. В связи с этим рост температуры системы способствует увеличению растворимости неметалла в металле. Растворение сопровождается ростом упругих деформаций, что приводит к увеличению расстояния между атомами металла в кристаллической решётке (т.е. росту параметров элементарной ячейки «хозяина»). Этот процесс происходит до тех пор пока внутреннее давление не достигнет критических значений, и не начнётся процесс упорядочения внедрённых атомов, который закончится формированием новой кристаллической структуры, характеризующий новый химический индивид – фазу внедрения. Например, тантал имеет кристаллическую структуру типа ОЦК, а в фазе внедрения состава TaC1-x атомы тантала располагаются по позициям ГЦК; скандий имеет структуру типа ГПУ, а в ScH2-x атомы скандия, также как в предыдущей фазе, располагаются по позициям ГЦК. Необходимо отметить, что образование фаз внедрения, в отличие от процесса формирования твёрдых растворов, приводит к уменьшению энтропии системы. Например, Но298 образования ZrH2-x и TiC1-x равны, соответственно, - 169,3 и – 183,5 кДж/моль, что свидетельствует об образовании между внедрёнными атомами и атомами металлов химических связей. Электроны внедрённых атомов в процессе образования новых связей изменяют электронную концентрацию в энергетических зонах, что, в соответствии с ранее рассмотренной концепцией, приводит к изменению строения кристаллической решетки исходной фазы «хозяина». (В рамках описания фаз внедрения, кристаллическую решётку металла предложено называть «хозяином», а внедрённые атомы – «гостями»).
Возможность образования фаз внедрения регламентируется правилом Хэгга: ra./r мe 0,59. Однако в данном случае учёт размерного фактора ( в отличие от фаз Курнакова и Лавеса) является необходимым, но недостаточным условием формирования соединений указанного типа. Показано, что эти фазы образуются только при средней или малой разности в ЭО атомов, образующих фазу и атомы «хозяина» должны обладать свойствами акцепторов.
Полученные в последние годы экспериментальные данные показывают, что фазы внедрения представляют собой системы со смешанным типом химической связи: делокализованной с участием, как электронов атомов металлов, так и электронов внедрённых частиц, локализованной, двухцентровой – неполярной между атомами одного элемента и полярной между атомами «гостя» и «хозяина», а также ионной. В частности, установлено, что атомы «гостей» в гидридах, карбидах и нитридах положительно поляризованы и в полях достаточной напряжённости перемещаются к катоду в виде катионов Эn+, что показывает недостаточность концепции электроотрицательности (согласно этой концепции ионизация более электроотрицательного атома в окружении менее электроотрицательных невозможна !). Доля различных типов связей в таких системах, естественно, зависит от общего числа валентных электронов и валентных орбиталей у взаимодействующих атомов. Когда у атомов «хозяина» достаточно свободных орбиталей для аккумулирования электронов внедрённых атомов, фазы внедрения характеризуются по сравнению с «хозяином» более высокими температурами плавления, твёрдостью, тепло- и электропроводностью, коррозионной устойчивостью, что указывает на значительное снижение в системе энергии Гиббса при их образовании. На основе зонной теории можно предсказать, что максимальный эффект внедрения будет наблюдаться в случае образования рассматриваемых фаз на основе электрон-дефицитных переходных элементов и лантаноидов. При этом максимальных значений этот эффект достигнет у элементов четвёртой и пятой побочных подгрупп, атомы которых являясь хорошими акцепторами, способны образовывать и достаточно большое число связей по обменному механизму. Например, как отмечалась выше для фаз внедрения температура плавления выше, чем у исходных металлов, однако в ряду фаз типа MeN1- x (где Ме = Ti, V, Cr) разность между температурой плавления нитрида и металла быстро уменьшается в связи с тем, что дефицит электронов у атомов «хозяина» снижается. Экстраполируя данные рассуждения на вторую половину рядов переходных элементов, можно сделать вывод, что образование соединений рассматриваемого типа проблематично для элементов подгруппы меди и невозможно для элементов подгруппы цинка. Предлагаем читателем попытаться применить концепцию, использованную при интерпретации фаз Лавеса, для подтверждения сформулированных выше выводов об изменении стабильности фаз внедрения при изменении числа электронов у атомов «хозяина» и «гостя».
Фазы внедрения обычно имеют плотноупакованную структуру типа ГПУ или ГЦК (к.ч.=12). Как известно в такой упаковке имеется два вида пустот – тетрагональные (Т), число которых в два раза больше числа атомов кристаллической решётки и октаэдрические (О) – их число равно числу атомов кристалла. В зависимости от размеров и с учётом правила Хегга, внедряемые атомы могут занимать либо пустоты Т , либо пустоты О, случаи, когда идёт одновременное заполнение пустот обоих видов, являются аномальными и на практике встречаются крайне редко. Очевидно, что если внедрение происходит только по позициям О, то предельный состав МХ, а её область гомогенности связана с возможным дефицитом в анионной подрешётке. Если же заполняются только тетраэдрические пустоты, то предельный состав фаз внедрения МХ2, а при заполнении пустот обоих видов МХ Также как для первого случая, изменение состава в этих соединениях возможно только в сторону снижения в них концентрации внедрённых частиц.
Таким образом, фазы внедрения можно рассматривать как новый этап в организации вещества, который контролируется не только размерным фактором, но и зависит от электронного строения, как атомов «хозяина», так и атомов «гостей». Это связано с тем, что в этих системах формируется тип химической связи, который трудно интерпретировать в рамках существующих теорий (теоретически это можно сделать в рамках «зонной» теории, однако количественные расчёты в этом методе ещё очень сложны, а приближения достаточно грубы). Многоцентровой с признаками локализации тип химической связи предопределяет лабильность рассматриваемых фаз по составу, изменением которого в пределах области гомогенности, можно плавно изменять их физические, химические, физико-химические, механические и электрофизические свойства, а следовательно, целенаправленно создавать материалы с заданными свойствами.
Подводя итог этой по рассмотренным бинарным фазам, проследим генетическую связь между отдельными формами, образующимися при взаимодействии переходных металлов с кислородом на примере системы Ti – О2. При низкой концентрации кислорода в этой системе на основе - Ti и - Ti образуются нерегулярные твёрдые растворы. Повышение концентрации кислорода в фазах твёрдых растворов способствует их упорядочению и трансформации в соединения Курнакова – сначала кластерного «(Ti2)3О», а затем классического типа «Ti3О». Дальнейший рост концентрации кислорода в фазах Курнакова вызывает их превращение в фазу внедрения Ti2О. Очевидно, что состав этой фазы предопределяется большим числом валентных электронов у атомов кислорода, а дальнейшее увеличение отношения О:Ti должно приводить к расщеплению единой зоны, т.е. к переходу в новое качество – полупроводниковую фазу. Средний состав этой фазы «TiО». Все названные фазы имеют значительные области гомогенности, так как гетеродейсмичный характер связи в них предопределяет формирование значительного числа макросостояний с близкой энергией. Окисление «TiО» приводит к образованию «Ti2О3» с узкой областью гомогенности (TiО1,49-1,51). Это объясняется ростом локализации химической связи в системе по мере уменьшения дефицита электронов в системе и увеличением ширины запрещённой зоны, по сравнению с предыдущими фазами. В свою очередь окисление «Ti2О3» приводит к формированию фаз типа TinО2n -1 (n = 4, 5, 6, 7, 8 …..) с блочной структурой и на последнем этапе наблюдается формирование TiО2, который отличается от предыдущих фаз минимальной областью гомогенности и максимальной величиной запрещённой зоны (диэлектрик).
Рассмотренные переходы показывают, как изменение количественного состава фаз постепенно изменяет их структуру, характер химической связи между атомами, свойства материалов на их основе, т.е. иллюстрирует закон перехода количества в качество. При этом с ростом концентрации кислорода в системе роль ЭО её атомов в формировании фаз постепенно увеличивается, а размерный фактор уходит на второй план. Одновременно увеличивается число и суммарная энергия химических связей в единице объёма системы, что резко повышает термодинамическую стабильность фаз (Go298 фаз рассмотренной системы изменяется от 20 -60 кДж/моль для твёрдых растворов и соединений Курнакова, до 250 - 310 кДж/моль – для «Ti2О3», TinО2n -1 и TiО2).
2.3 Галогениды, оксиды и сульфиды
Атомы галогенов кислорода и серы являются донорными, так как на валентном уровне имеют, соответственно, три и две неподелённые электронные пары. Следовательно, кроме связей, образованных по обменному механизму, в их соединениях могут образовываться ковалентные связи по донорно-акцепторному механизму, если партнёр по бинарному соединению имеет свободные орбитали. В свою очередь галогены и халькогены необходимо разделить на две части. Так атомы фтора и кислорода, не имеющие свободных орбиталей на валентном уровне могут быть только донорами, а атомы остальных элементов этих подгрупп, имеющие свободные орбитали валентного d-подуровня, могут (в зависимости от строения атома-партнёра) проявлять, как донорные, так и акцепторные свойства.
С точки зрения строения атома, стабильность бинарной фазы предопределяется соотношением суммарного числа валентных орбиталей атомов-партнёров к числу их валентных электронов. С точки зрения ММО ЛКАО, оптимальным в этом случае является соотношение близкое к 1:1, тогда электроны бинарной фазы заполняют все связывающие орбитали, что обеспечивает максимальное значение порядка связи. Очевидно, что, как при росте этого отношения, так и при его уменьшении порядок связи будет снижаться, что приведёт к дестабилизации рассматриваемых фаз.
При равном порядке связи у ряда форм, энергия связи (а также термодинамическая стабильность фазы) будет предопределяться её длинной, т.е. атомным (ионным) радиусом атома-партнёра.
В рамках указанных выводов рассмотрим ряды оксидов, сульфидов и галогенидов элементов третьего периода. В таблицах – 5 представлены значения Gо298 образования этих соединений, представленных в различных формах, для того, чтобы можно было бы их использовать для предсказания возможности протекания различных по характеру процессов.
Таблица Gо298 образования форм ЭОх и ЭхО
|
состав параметр |
NaO0,5 |
MgO |
AlO1,5 |
SiO2 |
PO2,5 |
SO3 |
ClO3,5 |
- |
|
- Gо298 |
190 |
570 |
792 |
857 |
675 |
371 |
- 155 |
- |
|
состав параметр |
Na2O |
MgO |
Al0,67O |
Si0,5O |
P0,4O |
S0,33O |
Cl0,29O |
|
|
- Gо298 |
380 |
570 |
528 |
428,5 |
270 |
124 |
- 44,3 |
|
|
к.ч.Эn+ |
4 |
6 |
6 |
4 |
4 |
4 |
4 |
- |
|
Z |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
- |
Z -число неподелённых электронных пар на 1 атом элемента
Таблица 4. Gо298 образования форм ЭSх и ЭхS
|
состав параметр |
NaS0,5 |
MgS |
AlS1,5 |
SiS2 |
PS2,5 |
- |
- |
- |
|
- Gо298 |
180,5 |
365 |
246 |
159 |
38,5 |
- |
- |
- |
|
состав параметр |
Na2S |
MgS |
Al0,67S |
Si0,5S |
P0,4S |
- |
- |
|
|
- Gо298 |
361 |
365 |
164 |
79,5 |
15,4 |
- |
- |
|
|
к.ч.Эn+ |
4 |
6 |
6 |
4 |
4 |
- |
- |
- |
|
Z |
1 |
2 |
3 |
4 |
5 |
- |
- |
- |
Z -число неподелённых электронных пар на 1 атом элемента
Таблица 5. Gо298 образования форм ЭClх и ЭхCl
|
состав параметр |
NaCl |
MgCl2 |
AlCl3 |
SiCl4 |
PCl5 |
PCl3 |
SCl2 |
SCl |
|
- Gо298 |
393 |
592 |
630 |
618 |
297 |
273 |
29 |
19 |
|
состав параметр |
NaCl |
Mg0.5Cl |
Al0,33Cl |
Si0,25Cl |
P0,2Cl |
P0,33Cl |
S0.5Cl |
SCl |
|
- Gо298 |
393 |
296 |
210 |
154,5 |
59.4 |
91 |
14.5 |
19 |
Из рассматриваемых элементов у атома натрия максимальное значение радиуса, восемь свободных орбиталей валентного уровня и один валентный электрон. В связи с этим в сульфиде и оксиде к.ч. этого атома низкое. ПД катиона натрия (в связи с максимальным значением радиуса, минимальным значением заряда и восьми электронной оболочкой) мало и, следовательно, у этой частицы не высокая акцептирующая способность. Небольшая доля -дативного взаимодействия в его оксиде и сульфиде предопределяется ещё и тем, что на один атом натрия в этих системах приходится только одна неподелённая электронная пара атома халькогена. Небольшое снижение стабильности формы при переходе от оксида к сульфиду связано, преимущественно, с большим радиусом атома серы, по сравнению с радиусом атома кислорода.
При движении по периоду слева направо радиус атомов и ионов элементов уменьшается, число свободных орбиталей у партнёров кислорода и серы уменьшается, а число атомов халькогенов, приходящих на один атом рассматриваемых элементов возрастает. Следовательно, по ряду слева направо, при неизменном числе орбиталей центрального атома, в системе растёт число неподелённых электронных пар (таб. и 4). На первом этапе это вызывает рост -дативного взаимодействия в рассматриваемых формах, что будет способствовать увеличению их стабильности. В случае оксидов оптимальное соотношение свободных орбиталей центрального атома и неподелённых пар атомов кислорода (5 на 4) у SiO2. У последующих оксидных фаз число свободных орбиталей центрального атома меньше числа неподелённых пар кислорода, следовательно, часть его электронной плотности атома кислорода не сможет принять участие в образовании химической связи и будет локализована вблизи него. Это вызовет резкий рост сил отталкивания в системах и снижение их термодинамической стабильности. Согласно же ММО ЛКАО в рассматриваемых оксидах фосфора, серы и хлора часть электронов будет находиться на разрыхляющих орбиталях, что объясняет снижение стабильности форм в указанном ряду, при этом Gо298 образования ClO3,5 будет уже положительной величиной.
Однако, необходимо помнить, что число связей в системе является только одним из критериев, по которым можно судить о стабильности той или иной фазы. Это связано с тем, что конечный вывод о суммарном изменении энергии в системе невозможно сделать без анализа энергии образующихся в ней химических связей, которая зависит от длины связи и типа орбиталей, участвующих в перекрывании. В частности, при одинаковом порядке связей в ЭОх и ЭSх максимум стабильности форм у элементов третьего периода смещается вправо, вопреки возможным прогнозам, основанным на понятии деформируемости и главном квантовом числе взаимодействующих орбиталей (таб. и 4). Напомним, что атомы серы, по сравнению с атомами кислорода, имеют бльшую деформируемость и электронные пары валентного уровня этого атома находятся на орбиталях, имеющих главное квантовое число равное трём, так же как и акцептирующие орбитали центральных атомов – эти характеристики, при прочих равных условиях, способствуют росту энергии -дативного взаимодействия.
Указанная «аномалия», преимущественно, связана с размерным фактором. Атом серы имеет орбитальный радиус на 30% больше, чем радиус кислорода, поэтому быстрое уменьшения радиуса центрального атома по периоду слева направо, приводит к росту отталкивания в системах, что способствует увеличению длины связей (при сохранении к.ч.) и уменьшению их энергии. Этим же объясняется понижение стабильности сульфидов по сравнению с оксидами при движении по периоду слева направо (табл.6).
Таблица 6. Изменение стабильности сульфидов по сравнению с оксидами для элементов третьего периода.
|
элемент параметр |
Na |
Mg |
Al |
Si |
P |
|
Gо298МехSy/Gо298Мех Oy |
0,952 |
0,640 |
0,311 |
0,185 |
0,124 |
На валентном уровне атомов галогенов, по сравнению с атомами халькогенов, на одну неподелённую пару больше и на один неспаренный электрон меньше. Это не только приводят к изменению соотношения атомов в бинарных соединениях при переходе от халькогенидов к галогенидам, но и способствуют более быстрому электронному насыщению галогенидных форм по периоду, по сравнению с ранее разобранными. Как следует из таблицы 5. стабильность хлорида натрия, при пересчёте на 1 моль катиона, практически в два раза выше, чем у оксида. Это связано с тем, что орбиталей у иона натрия вполне достаточно для аккумулирования электронных пар атомов хлора. Анализ данных таблицы 5 также показывает, что в периоде слева направо стабильность хлоридов в пересчёте на 1 моль ионов хлора снижается, а в пересчёте на 1 моль атомов центрального элемента, она примерно одинакова у хлоридов магния, алюминия и кремния, несмотря на рост числа -связей у хлоридов в указанном ряду и уменьшение радиуса центральной частицы. Как только число -связей прекращает увеличиваться (начиная с хлоридов фосфора), силы отталкивания в системе, вызванные избытком электронов, начинают в ней превалировать над силами, способными снижать энергию в системе, за счёт чего стабильность форм в рассматриваемом ряду резко падает при движении слева направо. Аналогичные результаты можно получить и исходя из ММО ЛКАО, – при фиксированном числе атомных орбиталей в системе, полное заполнение связывающих и несвязывающих орбиталей достигается тем быстрее, чем больше число валентных электронов у атомов, образующих систему.
Стабилизация электрон-избыточных галогенидов часто достигается при их полимеризации. Например, TiCl4 агрегатных состояниях мономерен (12 свободных орбиталей титана и 12 неподелённых пар атомов хлора, тогда как хлорид железа (III) димерен в газообразном и жидком состоянии, а в твёрдом образует псевдо молекулярную структуру, содержащую мостиковые атомы хлора. Аналогична ситуация и для других фаз рассматриваемого типа, при этом степень полимеризации, а также её конкретные формы будут зависеть, как от качественного и количественного состава галогенида, так и от параметров состояния системы (рис.9).
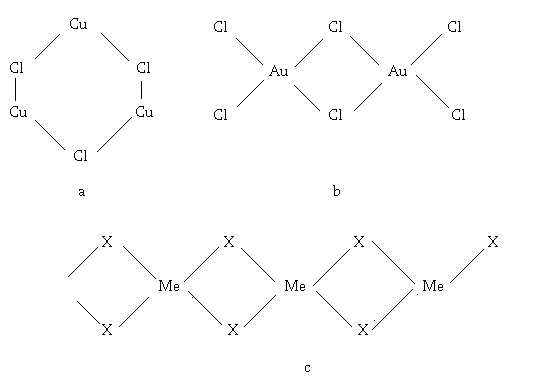
Рис.9. Структуры электрон избыточных галогенидов: а – (CuCl)3, b - Au2Cl6, c - (MeCl2)n (Me = Cu, Pd, X = Cl, Br, I).
При относительно небольшом избытке электронной плотности в системе в твёрдом состоянии происходит формирование слоистых решёток, которые состоят, фактически, из псевдо двумерных полимерных структур ( хлориды железа (II), кобальта (II), никеля (II), алюминия (III), иодид кадмия и т.д.).
В структурах многих низших галогенидов d-элементов обнаружены связи - Ме – Ме -. В ряде случаев такие бинарные группировки достаточно устойчивы даже в присутствии полярных растворителей, например – Hg – Hg - , стабильность которой предопределяется высокой деформируемостью орбиталей атомов ртути. В большинстве же случаев образуются группировки из нескольких атомов менее электроотрицательного элемента, связанные между собой ковалентной неполярной связью. Вещества, в состав которых входят группировки - Ме – Ме -, образующиеся, например, за счёт перекрывания d-, ds- или dsp-орбиталей атомов переходных элементов называются кластерными. Такие фазы формируются в случае электрон-дефицитных систем, т.е. когда электронов более электроотрицательного атома-партнёра недостаточно для образования -связей с участием всех валентных электронов металла. Стабилизация фаз данного типа возможна и за счёт образования дополнительных -связей по донорно-акцепторному механизму, например ниобий и тантал образуют галогениды состава: МеX1,83, МеХ2,33, МеХ2,5 и МеХ2,67, некоторые из них представлены на рисунках 10, 11 и 12. Указанные фазы состоят из кластерных катионов и внешнесферных анионов галогенов, при этом в зависимости от формальной степени окисления центрального элемента может меняться число внешнесферных анионов, но строение кластера при этом не изменяется. Поэтому сходную структуру катиона имеют фазы МеХ1,83 (Ме = Nb и Та) и МеХ2 (Ме = Mo, W, Pt и другие элементы второго и третьего переходного рядов). В рассматриваемых частицах ионы галогенов занимают вершины куба, а атомы переходных элементов – центры граней этого куба, образуя октаэдрический по форме кластер типа Ме6. Внешнесферные ионы галогенов, например, в [Nb6X8]Х3 и [Mo6X8]Х4, образуют мостики, соединяющие кластеры между собой.
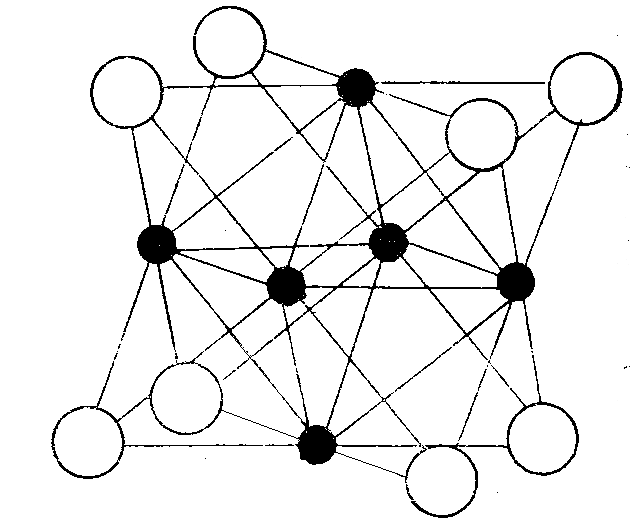
Рис.10. структура ионов [Nb6X8]3, [Mo6X8]4,+ [Pt6X8]4+ и т.д. (Х = Cl, Br, I).
Тёмные шары – атомы металла, светлые – атомы галогенов.
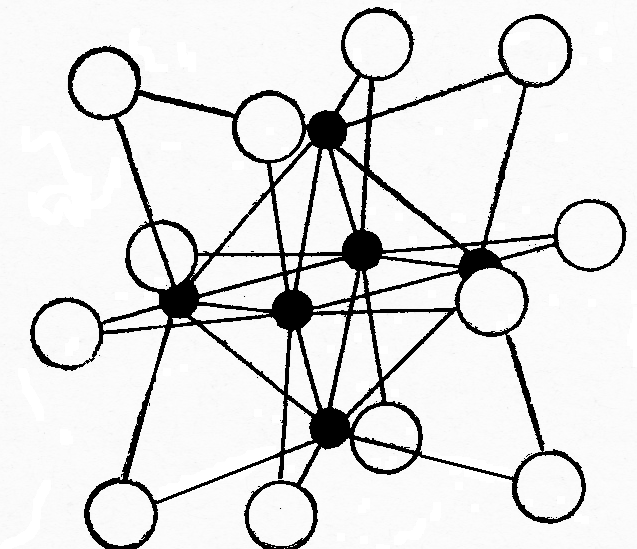
Рис.11. структура ионов [Ме6X12 ]2+, (где Ме = Nb и Та, Х = Cl, Br, I). Тёмные шары – атомы металла, светлые – атомы галогенов.
В ионе [Ме6X12 ]2+ ионы галогенов занимают центры рёбер куба, в который вписан октаэдрический кластер из атомов Nb или Та. Роль внешнесферных ионов такая же, как в предыдущей форме.

12. Структура МеХ2,67 (где Ме = Nb и Та, Х = Cl, Br, I). Тёмные шары – атомы металла, светлые – атомы галогенов.
В структуре МеХ2,67 (где Ме = Nb и Та, Х = Cl, Br, I) экспериментально установлено образование кластеров, состоящих из трёх атомов Nb или Та, расположенных в одной плоскости.
Анализ полученных по кластерным формам данным приводит к выводу, что число атомов в кластере уменьшается с ростом в системе концентрации электроотрицательного атома. Кластеры подобного типа были обнаружены и в структурах низших оксидов переходных элементов, тогда как для сульфидов при низкой степени окисления катионов чаще наблюдается образование кластерных форм серы, которые создают условия для формирования полисульфидов.
Роль неподелённых электронных пар в бинарных соединениях галогенов и халькогенов просматривается, как при анализе стабильности соединений d-элементов в периодах и подгруппах, так и при рассмотрении изменении стабильности однотипных фаз в главных подгруппах (таб.8). Например, для имеющих одинаковую кристаллическую структуру оксидов d-элементов первого переходного ряда, несмотря на тенденцию к уменьшению в ряду радиусов ионов Ме2+, Но298 образования их оксидов растёт, а энергия атомизации оксидов снижается (таб.7).
Таблица 7. Но298 образования и энергия атомизации оксидов типа МеО
|
Ме параметр |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
|
- Но298 кДж/моль |
526 |
432 |
389 |
361 |
265 |
239 |
219 |
162 |
|
Еат. кДж/моль |
1241 |
1187 |
1052 |
975 |
928 |
913 |
911 |
738 |
Очевидно, что выявленные закономерности связаны с ростом числа валентных электронов у атомов d-элементов, при этом резкий рост Но298 образования рассматриваемых форм происходит при переходе от оксида марганца к оксиду железа, что связанно с появлением у атома железа неподелённой электронной пары на 3d подуровне. В подгруппах d-элементов наблюдается стабилизация однотипных форм оксидов, в связи с ростом энергии химической связи в системе за чёт увеличения деформируемости электронных орбиталей атомов и повышения к.ч. центрального катиона.
Таблица 8. Gо298 образования диоксидов элементов подгруппы углерода и к.ч. в них центрального атома
|
состав параметр |
CO2 |
SiO2 |
GeO2 |
SnO2 |
PbO2 |
|
Gо298 кДж/моль |
- 365 |
- 857 |
- 522 |
-520 |
- 220 |
|
к.ч. |
2 |
4 |
4 |
6 |
6 |
В главных подгруппах, при переходе от первого элемента ко второму, у атома последнего появляются пять орбиталей валентного d-подуровня, что приводит к резкому увеличению доли -дативного взаимодействия в системе и росту к.ч. центрального атома. Оба эти фактора способствует резкому снижению Gо298 образования однотипных по составу форм в указанном направлении. Строение и заселённость зон в SiO2 и GeO2 одинаковы, однако валентными у германия являются орбитали уровня с большим значением главного квантового числа, а его радиус больше радиуса атома кремния. Указанные изменения в системе приводят к снижению суммарной энергии связи в системе и росту значения Gо298 образования. При переходе от GeO2 к SnO2 энергия единичной связи Э – О уменьшается за счёт выше указанных причин, но рост числа связей в системе компенсирует рост энергии системы за счёт первых двух факторов. В результате Gо298 образования двух рассматриваемых форм, практически, одинакова. Надеемся, что рост значений Gо298 образования при переходе от SnO2 к PbO2, читатель сможет объяснить самостоятельно.
Кроме фактора оптимального соотношения валентных электронов и валентных орбиталей в системе и размеров образующих систему частиц на стабильность бинарных форм оказывает влияние деформируемость орбиталей, принимающих участие в образовании химической связи (или в соответствии с ММО ЛКАО эффект дополнительного снижения энергии МО). Эту зависимость можно рассмотреть на примере высших оксидов d-элементов, с высокой долей ковалентной составляющей химической связи. Известно, что радиусы атомов и ионов (в одной и той же степени окисления) в подгруппах d-элементов изменяются мало за счёт d- и f-сжатия. Следовательно, изменение Gо298 образования и энергии атомизации (в первом приближении) в этом случае можно проследить без учёта размерного фактора (таб.9.).
Таблица 9. Gо298 образования и энергии атомизации триоксидов подгруппы хрома
|
состав параметр |
CrO3 |
MoO3 |
WO3 |
|
Gо298 кДж/моль |
- 507 |
- 678 |
- 763 |
|
Еат. кДж/моль |
1428 |
1755 |
1892 |
Как следует из представленных данных, рост деформируемости электронных орбиталей, а также увеличение к.ч. Ме6+ в подгруппе при переходе от хрома к молибдену, приводит к значительной стабилизации оксидов.
Описанная теоретическая концепция позволяет, не прибегая к сложным расчётам, ориентироваться в материале, относящемуся к бинарным фазам. Рассмотрим возможности данного подхода на нескольких примерах.
1. Оценим термодинамическую возможность самопроизвольного превращения оксидов элементов третьего периода в пероксиды.
Согласно сформулированным выше выводам Na2O является электрон-дефицитным веществом, при этом у двух ионов натрия только низко лежащих s- и p-орбиталей – восемь, а общее число свободных орбиталей валентного уровня – восемнадцать. Число же валентных электронов на формульную единицу – восемь (два электрона атомов натрия и шесть электронов атомов кислорода), следовательно, четыре низко лежащих sp-орбиталей – свободны. Тогда присоединение к системе ещё одного атома кислорода должно привести к её стабилизации: Na2O + 1/2O2 = Na2O2. Проверим вывод с помощью расчёта: согласно табличным данным Gо298 этой реакции (-448 + 380 = -68 кДж/моль), т.е. она действительно возможна ( и реально протекает) при с.у..
В случае MgO при том же числе электронов валентных уровней, число валентных орбиталей катиона в два раза меньше. Так как в данном случае все низко лежащие s- и p-орбитали атома магния уже принимают участие в образовании химических связей с атомом кислорода, то для формирования пероксида этого элемента необходимо дополнительное привлечение ещё четырёх из пяти свободных d-орбиталей центрального катиона. Следовательно, согласно выводам рассматриваемой концепции, образование MgO2 по реакции: MgO + 1/2О2 = MgO2 теоретически возможно, но менее вероятно чем превращение Na2O в Na2O2. Для анализа воспользуемся термодинамическим расчётом изменения функций состояния для обсуждаемого процесса: Но298 = - 623,0 + 602,2 = - 20,8 кДж/моль; Sо298 = (85,8 – 26,9 – 102,6)·10-3 кДж/моль = - 43,7·10-3 кДж/моль, тогда Gо298 этой реакции: - 20,8 – (- 43,7·10-3·298) = - 7,77 кДж/моль. Таким образом, с термодинамической точки зрения, этот процесс возможен при с.у.. Однако у MgO очень высокая энергия кристаллической решётки и, следовательно, при с.у. Еак. данного процесса достигнута не будет. Повышение же температуры системы будет смещать равновесие процесса влево в соответствии с сочетанием изменения энтальпии и энтропии (Но < 0, Sо < 0). Таким образом, получение MgO2 из MgO невозможно в связи с кинетическими ограничениями.
Читателю предлагается самостоятельно доказать, что образование пероксидов алюминия, кремния, фосфора, серы и хлора, в процессе их взаимодействия с кислородом невозможно, в связи с недостаточным числом свободных валентных орбиталей у центральных атомов.
2. Оценим термодинамическую возможность восстановления оксидов представленных в виде ЭхО (таб.) простыми веществами того же ряда элементов:
ЭхО + yЭ = Эy О + хЭ
В связи с тем, что в данном ряду минимальное значение Gо298 образования имеет MgO, с помощью Mg может быть восстановлен любой из оксидов данного ряда. В промышленности этот способ применяется для получения натрия и кремния: Si0,5O + Mg = MgO + Si (Gо298 = - 528 + 429 = = -99 кДж/моль. Необходимо напомнить, что в процессах указанного типа протекают последовательные процессы, поэтому получаемое вещество в качестве примесей содержит алюминиды, силициды, фосфиды и сульфиды магния, а в системе Cl2O7 процесс развивается со взрывом уже при с.у. и приводит к образованию не только MgO и Cl2, но и MgCl2 , а также О2. Читателю предлагается ответить на вопрос о возможности протекания аналогичных процессов при использовании в качестве прекурсоров галогенидных фаз.
Установим, как на основе понятия «термодинамическая стабильность», связанной, как было показано выше с концепцией «неподелённых электронных пар» сделать прогноз возможности синтеза простых веществ галогенов, путём окисления их соединений молекулярным кислородом, исходя из следующих табличных данных: Gо298 образования в кДж/моль форм Al0,67O, Al0,33F и Al0,33Cl равны, соответственно: -528, -477 и -210. Тогда: 2Al0,33Cl + 1/2O2 = Al0,67O + Cl2
(Gо298 реакции = -528 + 2·210 = -108 кДж) т.е. Cl2 можно получать окислением хлорида алюминия на воздухе, при этом рост температуры будет способствовать протеканию процесса не только с кинетической, но и с термодинамической точки зрения, так как прямой процесс имеет положительный знак изменения энтропии.
Однако процесс: 2Al0,33F + 1/2O2 = Al0,67O + F2 термодинамически невозможен (в чем мы предлагаем убедиться читателям), тогда попытаемся в эту электрон-избыточную систему ввести частицы, имеющие достаточно большое число свободных орбиталей, и тем самым снизим суммарную энергию системы:
3Al0,33F + Al + 3/2O2 = 3Al0,67O + 3/2 F2
(Gо298 реакции = - 3·528 + 3·477 = - 153 кДж), т.е. F2 можно получать окислением фторида алюминия на воздухе в присутствии алюминия, при этом рост температуры системы с точки зрения термодинамики не будет препятствовать протеканию прямого процесса, т.к. изменение энтропии в этом случае не велико.
4. Кислотно-основные свойства бинарных соединений галогенов и халькогенов в значительной степени связаны с дефицитом или избытком электронов в системах, в том понимании, которое придаёт данному термину рассматриваемая концепция. Как уже отмечалось, причина низкой стабильности бинарных фаз может быть связана, как со значительным превышением числа валентных электронов системы над числом валентных орбиталей, так и с малым числом валентных электронов по сравнению с числом валентных орбиталей. Следовательно, энергетически выгодным будет процесс взаимодействия между «электрон-дефицитными» и «электрон-избыточными» фазами. Первые называются – основными, а вторые – кислотными. Очевидно, что фазы, в которых число валентных орбиталей будет близко к числу валентных электронов, называются амфотерными.
Тогда следующий этап организации вещества фактически сводится к устранению недостатков, связанных с избытком или недостатком валентных электронов в системах, при котором обе исходные формы переходят в более стабильное состояние, образуя новый индивид. Рассмотрим несколько процессов, в которых один моль Na2O взаимодействует с оксидами элементов третьего периода:
Na2O + Al2O3 = 2NaAlO2 (Gо298 = - 173 кДж/моль)
Na2O + SiO2 = Na2SiO3 (Gо298 = -194 кДж/моль)
Na2O +1/6P4O10 = 4/6Na3PO4 (Gо298 = -387 кДж/моль)
Na2O + SO3 = Na2SO4 (Gо298 = -552 кДж/моль)
Na2O + Cl2O7 = 2NaClO4 (Gо298 = -570 кДж/моль)
Очевидно, что эффективность процессов в пересчёте на один моль «электрон-дефицитного» оксида Na2O растёт по мере увеличения числа избыточных неподелённых пар у партнёра.
5. Какие методы позволяют стабилизировать соединения галогенидов?
Стабилизация галогенидных фаз, также как оксидных или сульфидных возможна за счёт введения в систему частиц с достаточно большим числом валентных орбиталей. В результате этого, достаточно часто, происходит увеличение к.ч. исходного катиона, по отношению к ионам галогенов, что свидетельствует о второстепенной роли размерного фактора при формировании форм, в том случае, когда размер катиона соизмерим с размером катиона. Например, Gо298 образования PbCl4 больше нуля, т.е. эта фаза самопроизвольно разлагается при с.у.. В то же время Gо298 процесса:
PbCl2 + Cl2 + 2KCl = K2[PbCl6] имеет Gо298 < 0
и комплексный ион [PbCl6]2- достаточно устойчив не только при умеренно высоких температурах, но и в присутствии полярных растворителей; считается, что MoCl6 не может быть получен из за размерного фактора, поэтому при окислении молибдена хлором предельной формой является MoCl5, а при недостатке хлора в системе образуются рассмотренные выше кластерные структуры. В то же время в результате процесса:
Mo + 3KCl + 3/2Cl2 = K3[MoCl6]
образуется устойчивый комплексный анион, с таким же к.ч., как в MoCl6. Очевидно, что различия между метастабильными и стабильными формами в этих системах заключаются в большем числе свободных орбиталей в последних системах, за счёт наличия в них ионов калия, имеющего большое число валентных орбиталей. Читателю предлагается ответить на вопрос, будет ли столь же эффективным использование в описанных процессах хлоридов натрия и лития?
6. Как галогениды различных элементов будут относиться к воде?
Как отмечалось выше, термодинамическая стабильность по периоду для галогенидных фаз (в пересчёте на 1моль центрального атома) падает быстрее, чем оксидных. Отсюда можно сделать вывод, что галогениды всех р- и d-элементов будут гидролизоваться по катиону, а из галогенидов s-элементов – только галогениды бериллия и магния. Очевидно, что чем больше отличается стабильность галогенида от стабильности оксида (гидроксида) тем выше скорость данного процесса и его глубина. Поэтому, если хлориды магния или алюминия гидролизуются (в зависимости от условий проведения процессов) до оксо- или гидроксосолей (основные соли), то галогениды кремния или фосфора гидролизуются на 100%. Очевидно, что при переходе от галогенидных форм к оксидным на один атом центрального элемента приходиться в два раза меньше атомов-партнёров и у кислорода на валентном уровне на один валентный электрон меньше, чем у атомов галогенов. Следовательно, замена галогена на кислород энергетически выгодна для большинства галогенидов.
Аналогичный подход позволяет объяснить, почему галогениды магния гидролизуются, а галогениды щелочноземельных элементов нет. Напомним, что при переходе от магния к кальцию, у последнего резко возрастают валентные возможности за счёт появления у его атома пяти свободных орбиталей d-подуровня. В связи с этим стабильность его галогенидов возрастает практически в два раза: Gо298 образования MgCl2 составляет -592,2 кДж/моль, а Gо298 образования CaCl2 = -1011,5 кДж/моль, тогда как изменение Gо298 образования гидроксидов значительно меньше:-834,3 и -899,2 кДж/моль, соответственно. Предлагаем читателю убедиться в правоте выводов, рассчитав Gо298 гидролиза по катиону для ионов магния и кальция.
3 Гидроксиды и неорганические полимеры
3.1. Обзор строения и свойств гидроксидов различных элементов
Свойства и строение гидроксидов предопределяется их составом, т.е. строением входящих в них атомов, их радиусом и поляризующим действием центрального катиона.
Гидроксиды элементов главной подгруппы первой группы являются преимущественно ионными фазами, достаточно хорошо растворимыми в воде и имеющие истинную диссоциацию в этих растворах, равную 100%. С ростом ПД (поляризующего действия) центрального катиона полярность его связи с атомом кислорода группы ОН будет уменьшаться, а полярность связи О – Н – увеличиваться, что приведёт к следующим изменениям в системах: вещества будут изменять строение от ионного к ионно-ковалентному, затем произойдёт стабилизация полимерных форм, в которых степень полимеризации с ростом ПД будет постепенно снижаться и, наконец, образуются островные (молекулярные) структуры, характерные для гидроксидов с максимальным значением ПД центрального катиона. Одновременно с этим будут меняться кислотно-основные свойства рассматриваемых фаз: от типично основных (при минимальном значении ПД), через амфотерные (при среднем значении ПД) к типично кислотным (при максимальном значении ПД).
В рядах гидроксидов одного и того же элемента, с ростом поляризующего действия центральной частицы в анионах одноосновных кислородных кислот, их степень диссоциации будет расти. Например:
По мере увеличения степени окисления ионов Cln+ их радиус уменьшается, т.е. оба фактора (уменьшение радиуса ионов и рост их заряда) увеличивают поляризующее действие центральной частицы в данном ряду слева направо. Это ведет к увеличению полярности связи Н–О, уменьшению ее энергии и, как следствие, к росту степени диссоциации кислот Кд1 (НСlO) = 5·10–8, Kд1(HСlO2)=2·10–2, HClO3 и HСlO4 – сильные электролиты. По мере сближения поляризующего действия частицы, соединенной с атомом кислорода группы ОН–, с поляризующим действием иона Н+, значения констант диссоциации соединений типа R–О–Н по кислотному и основному типу сближаются. Такие электролиты, как известно, называются амфотерными. Если же поляризующее действие Rn+ в соединении типа R(ОН)n значительно ниже, чем у иона водорода, в наибольшей степени гидратируется Rn+, что способствует росту вероятности диссоциации формы по основному типу. Например, в ряду гидроксидов хрома, условно записанных в виде:
поляризующее действие ионов Crn+ возрастает (за счет роста их зарядов и уменьшения радиусов), что способствует увеличению значений констант диссоциации гидроксидов по кислотному типу: «Cr(OH)2» – основание, «Cr(OH)3» – амфотерный электролит, H2СrO4 – сильная кислота.
В малых периодах слева направо свойства гидроксидов, в которых атомы элементов находятся в высшей положительной степени окисления, изменяются от типично основных через амфотерные к кислотным (табл.10).
Таблица 10. Характер гидроксидов и их константы диссоциации по первой (К1) и второй (К2) ступеням.
|
Состав |
NaOH |
«Mg(OH)2» |
«Al(OH)3» |
H4SiO4 |
H3PO4 |
H2SO4 |
HClO4 |
|
Характер гидроксид. |
сильное основание |
слабое основание |
амфотерный электролит |
слабая кислота |
средняя кислота |
сильная кислота |
сильная кислота |
|
К1 |
4·10–1 |
2·10–6 |
1,3·10–10 |
7,6·10–3 |
|||
|
К2 |
3·10–3 |
4·10–13 |
1,6·10–12 |
6,2·10–8 |
1,2·10–2 |
С точки зрения теории поляризации такое изменение объясняется тем, что слева направо поляризующее действие ионов Na+–Mg2+–Al3+–Si4+–P5+–S6+–Cl7+ увеличивается за счет уменьшения их радиуса и роста заряда при сохранении 8е оболочки.
В главных подгруппах наблюдается общая тенденция к увеличению сверху вниз основных свойств гидроксидов (при одинаковой степени окисления центрального иона). Это обусловлено увеличением радиусов ионов этих элементов, что приводит к снижению их поляризующего действия (табл. 11).
Таблица 11. Константы диссоциации гидроксидов по основному типу
|
Состав |
«Be(OH)2» |
«Mg(OH)2» |
Ca(OH)2 |
Sr(OH)2 |
Ba(OH)2 |
|
К2 |
5·10–11 |
3·10–3 |
4·10–2 |
9·10–2 |
7·10–1 |
Однако, при прогнозе изменения значений констант диссоциации по основному (кислотному) типу, в данном случае необходимо учитывать возможность изменения электронной конфигурации иона. Например:
H3PO4 H4SiO4 H2SO4
К1 7,6·10–3 К1 1,3·10–10 К2 1,2·10–2
Эл. конф. иона 8е 8е 8е
H3AsO4 H4GeO4 H2SeO4
К1 6,0·10–3 К1 1,1·10–10 К2 9·10–3
Эл. конф. иона 18е 18е 18е
Близость Кд гидроксидов элементов одной и той же подгруппы связана с компенсацией фактора, уменьшающего поляризующее действие ионов (рост радиусов), фактором, увеличивающим их поляризующее действие (изменение электронной конфигурации от 8е к 18е).
Значительное влияние на степень диссоциации гидроксидов оказывает и их химическое строение, например:
H3P3O9 (HPO3)3 H4P2O7 H3PO4
K1 1,14 3,0·10–2 7,6·10–3
К2 2·10–1 4,4·10–3 6,210–8
К3 9·10–3 2,5·10–7 4,410–13
В молекулах всех трех представленных кислот степень окисления фосфора +5, т.е. его поляризующее действие формально одинаково, что может без учета фактора строения этих веществ может привести к неверному выводу о близких значениях Кд всех этих форм. На рис.13 представлены графические формулы молекул рассматриваемых соединений.
Рис.1 Графические формулы фосфорных кислот
Из этого рисунка следует, что причиной изменения Кд этих кислот является изменение их строения, сопровождающееся уменьшением их основности и, следовательно, снижающее роль контрполяризующего действия ионов водорода, что пропорционально росту ПД центрального иона. Другими словами за счёт высокой основности молекулы Н3РО4 полярность связи Н–О в ней невелика, что предопределяет относительно низкие значения ее даже первой Кд. В H4P2O7 на каждый ион фосфора приходится только по две гидроксогруппы, что увеличивает полярность связи Н – О и росту её Кд. То же относится и к H3P3O9, но так как изменение числа групп Н–О, приходящихся на один ион фосфора уменьшается в два раза (а не в 1,5 раза как в предыдущем случае), изменения в значении Кд будут ещё больше (рис.13).
Последовательная ионизация многоосновных кислот и многокислотных оснований также приводит к изменению ПД центральной частицы. При отщеплении иона водорода от группа –ОН, отрицательный заряд на ионе кислорода возрастает, что увеличивает его деформируемость. Это, в свою очередь, приводит к снижению ПД центральной частицы и уменьшению деформации ионов кислорода в оставшихся в анионе группах О-Н. Как было показано выше, это уменьшает значения Кд по кислотному типу:
H3PO4 H+ + H2PO4– K1=7,6 · 10-3
H2PO4– H+ + HPO42– K2=6,2 · 10-8
HPO42– H+ + PO43– K3=4,4 · 10-13
При диссоциации по основному типу отщепление группы ОН– от гидроксида увеличивает ПД катиона и полярность связи между катионом и анионами кислорода оставшихся групп ОН уменьшается, что затрудняет процесс диссоциации по данному типу. Например, для «Pb(OH)2» константы диссоциации по основному типу составляют: К1=9,5·10–4, К2=3,0·10–8.
Как уже отмечалось ранее, поляризующее действие ионов и их взаимная деформируемость предопределяет не только преимущественный характер диссоциации соединений типа R–О–Н и глубину протекания процесса их ионизации, но и состав этих соединений. Рассмотрим данный вопрос на примере гидроксидов элементов III периода Периодической системы. Например, теоретически гидроксид Р(V) может иметь состав Р(ОН)5. Однако за счет высокого поляризующего действия ионов Р5+ происходит значительная односторонняя деформация электронной плотности анионов кислорода групп ОН, что должно способствовать уменьшению длин связей Р–О–.Однако, сближение групп О-Н с центральной частицей приводит к увеличению отталкивания между ними. Противодействие сил отталкивания и притяжения способствует тому, что часть связей Р–О действительно укорачивается, а другие связи удлиняются:
Уменьшение длины и полярности связи между ионом фосфора и кислорода, как было показано выше, облегчает отрыв иона Н+, тогда как с ростом длины связи Р–О возрастает вероятность диссоциации соединения по основному типу. В результате этого в системе протекает процесс "внутримолекулярной нейтрализации", сопровождающийся образованием молекулы Н2О. Атом кислорода, оставшийся у иона фосфора после отщепления молекулы Н2О, еще в большей степени деформируется центральным ионом. Это способствует образованию между этими частицами дополнительной связи и снижению ПД иона фосфора. В связи с уменьшением ПД центральной частицы, деформация оставшихся 3-х групп ОН уменьшается, что снижает вероятность дальнейшего протекания процесса, связанного с уменьшением основности кислоты. В тоже время, при ещё бльшем ПД центральной частицы становятся возможными и последующие его стадии:
Таким образом, можно сделать вывод, что с ростом ПД центрального иона основность кислот будет уменьшаться. (В приведенном примере ионы Р5+, S6+, Cl7+ имеют одинаковое строение электронных оболочек, а их радиусы по Полингу равны r(Р5+)=0,34, r(S6+)=0,29, r(Cl7+)=0,26, что с учетом роста зарядов ионов резко увеличивает ПД по ряду слева направо.)
Ион Si4+ по сравнению с Р5+ характеризуется меньшим ПД, поэтому одной из форм кремниевых кислот является H4SiO4 – ортокремниевая кислота, существующая только в очень разбавленных водных растворах. Снижение ПД центрального иона приводит к сближению значений Кд этого гидроксида по кислотному и основному типу. За счет этого между молекулами данного гидроксида протекает процесс поликонденсации ("межмолекулярной нейтрализации"):
Так как появление атома кислорода между двумя атомами кремния влияет на значения ПД центральных частиц, дальнейший процесс поликонденсации приводит к образованию многочисленных полимерных форм кремниевых кислот линейного, слоистого или объемного строения. С ростом степени поликонденсации растворимость образующихся соединений уменьшается, и они выделяются из раствора в виде геля или же образуют с жидкой фазой коллоидный раствор.
При уменьшении заряда катиона на единицу при сохранении электронной конфигурации иона (Al3+), Кд по кислотному и основному типу остаются достаточно близкими ( в отличие от H4SiO4 Кд гидроксида Al(III) по основному типу больше, чем Кд по кислотному). Это, как мы видели на примере H4SiO4, должно приводить к формированию полимерной структуры этого вещества и выделению его из раствора в виде гелеобразной фазы. Такая же ситуация сохраняется и в случае гидроксида Mg(II), когда Кд осн > Кд кис, однако степень поликонденсации в этом случае значительно ниже, чем в случае гидроксида Al(III). Это, в частности приводит к тому, что растворимость гидроксида Mg(II) в воде в среднем в 1000 раз выше, чем у гидроксида Al(III). При дальнейшем снижении ПД катиона процесс поликонденсации подавляется, поэтому наличие полимерных частиц в растворах NaOH или Ca(OH)2 не фиксируется. Отсюда следует важный в практическом отношении вывод:
Одноядерные формы гидроксидов кислотного типа образуются только в случае достаточно высокого ПД центральной частицы аниона. Эти формы хорошо растворимы в Н2О и имеют высокие значения степени электролитической диссоциации.
При среднем ПД катиона образуются полимерные формы гидроксидов, плохо растворимые в воде и имеющие низкие значения Кд как по основному, так и по кислотному типу.
Гидроксиды катионов, характеризующихся низким ПД, представляют собой кристаллические вещества с преимущественно ионным характером связи. Они хорошо растворимы в воде и их степень диссоциации по основному типу равна 100% (по первой стадии в случае двукислотных гидроксилов).
В заключение отметим положительные и отрицательные стороны рассмотренной теории. Ценность любой теории оценивается с точки зрения степени согласования ее выводов с экспериментальными данными. Как следует из выше изложенного материала, теория поляризации дает правильный прогноз химических и физико-химических свойств разнообразных веществ, предсказывает их строение и состав. Однако выводы этой теории скорее качественные, чем количественные. Например, с ее помощью можно показать, что ВеО·хН2О является амфотерным гидроксидом и Кд его по основному и кислотному типу имеют низкие значения. Однако она не позволяет рассчитать абсолютные величины этих констант. С другой стороны, успешно интерпретируя изменение свойств соединений с одинаковой электронной структурой (по периодам и подгруппам), теория поляризации сталкивается со значительными трудностями при сравнении строения и свойств соединений, в состав которых входят ионы различных подгрупп с одинаковым строением электронных оболочек. Например, ионы Cr6+ и S6+ имеют 8е оболочку, r(Cr6+)=0,52, r(S6+)=0,29, следовательно ПД S6+ выше ПД Cr6+, что подтверждается значениями К2 для H2SO4 и H2CrО4, которые равны, соответственно, 1,2·10–2 и 3,2·10–7, и более низкой термической H2CrО4 устойчивостью по сравнению с H2SO4. Но тогда с точки зрения теории поляризации основность хромовой кислоты должна быть выше, чем серной, что не подтверждается на опыте. Например, дихромовая кислота могла бы иметь строение:
тогда как экспериментально установлено, что она двухосновна:
Рассчитать, какая же из представленных форм для дихромовой кислоты энергетически более выгодна, теория поляризации не позволяет. Однако, несмотря на указанные недостатки, рассмотренная теоретическая концепция дает возможность ориентироваться в разнообразном фактическом материале неорганической химии без использования сложных математических расчетов, что выгодно отличает ее от квантовомеханических теорий химической связи.
Так как большинство известных гидроксидов имеет в своём составе катионы со средним поляризующим действием, можно утверждать, что самым распространённым типом неорганических гидроксидов являются полимерные формы различного строения и переменного состава. Рассмотрим подробно строение и свойства таких гидроксидов на примере фаз ЭО2·хН2О (Э = Ti, Zr, Sn). Выбор этих форм вызван тем, что они являются типичными представителями своего класса соединений и переход к пониманию строения и состава других подобных форм, содержащих катионы других элементов, может быть осуществлён без существенного изменения основных понятий и положений.
3.2. Способы синтеза фаз с условной формулой ЭO2xH2O
В связи с высокой энергией кристаллических решёток ЭO2 (Э = Ti, Zr, Sn) их гидроксиды (IV) могут быть получены только косвенным путем. В частности, ЭO2xH2O образуются при взаимодействии растворов ацидокомплексов Э(IV) с раствором аммиака, а также в процессе кислотного гидролиза титанатов, цирконатов и станатов щелочных металлов. Установлено, что состав и свойства гелеобразных осадков, получаемых при проведении указанных процессов, зависит от концентрации исходных растворов, природы аниона, природы осадителя, температуры системы и т.д.. Содержащие минимальную концентрацию примесей, активные, рентгеноаморфные формы этих гидроксидов можно получить низкотемпературным гидролизом тетра-изо-пропилатов данных элементов. Температура системы во время синтеза не должна превышать 10 С. После промывания осадка методом декантации и высушивания при с.у., содержание в них воды составляет от 40 до 80 масс. %, что соответствует формуле ЭO2(3 – 9)Н2O.
Золи этих гидроксидов можно получить электролизом водных растворов H2[ЭCl6]. Сущность метода состоит в удалении из раствора ионов H+ и Cl- за счёт протекания анодного и катодного процессов, что способствует увеличению степени гидролиза исходных форм и удалению анионов из системы. Процесс осуществляется в трехкамерном электролизере с использованием ионообменных мембран. Электролиз 1М водного раствора H2[ЭCl6] проводят при температуре порядка 40С. Протекающий процесс гидролиза способствует формированию многоядерных оловых форм гидроксида Э(IV), с последующим их переходом в -форму, которая на следующем этапе разлагается. При достижении системой атомного соотношения Cl-/Э4+ 2 наблюдается выпадение мелкодисперсного осадка ЭO2, имеющего, по данным РФА, в случае диоксидов титана и олова искажённую структуру типа рутила (рис.14)., а в случае диоксида циркония смесь тетрагональной и моноклинноц модификаций. Для получения фаз с гораздо меньшим атомным отношением Cl-/Э4+ необходимо введение стабилизирующей добавки, в качестве которой может быть использован хлорид циркония. Добавка этого реагента в систему в количестве 1 мол. % позволяет получить золь с атомным отношением Cl-/Э4+ = 0,22 - 0,25.
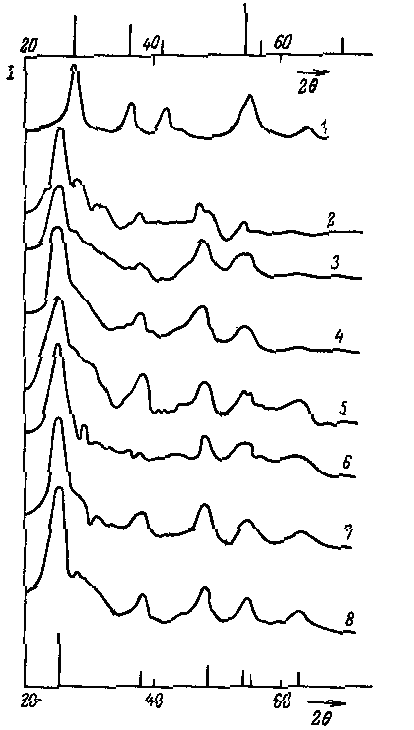
Рис. 14. Дифрактограммы образцов: 1-осадок продукта гидролиза H2[TiCl6], полученный без стабилизатора; осадков продуктов гидролиза H2[TiCl6], легированных 1 моль. % соединений Zr(IV) при мольном соотношении Сl-/Ti4+ в образцах: 2-1,2; 3-1,01; 4-0,63; 5-0,3 Осадков продуктов гидролиза H2[TiCl6], легированных 5 моль. % соединений Zr(IV) при мольном соотношении Сl-/Ti4+ в образцах: 6-0,75; 7-0,63; 8-0,45.
Части штрихрентгенограмм: верхняя - рутил, нижняя – анатаз
Установлено, что стабилизирующее действие соединений циркония обусловлено увеличением заряда и изменением природы поверхности гранул коллоидных частиц, вследствие сорбции на них положительно заряженных циркониевых гидроксокомплексов. По мере уменьшения атомного отношения Cl-/Э4+ в рассматриваемой системе начинается процесс кристаллизации ЭO2. Кристаллические частицы, в основном, имеют указанную выше структуру и размеры порядка 100 (в этих условиях возможно образование и кристаллического TiO2 со структурой анатаза).
Фазовый состав продуктов гидролиза указанных соединений переходных и непереходных элементов зависит от состава прекурсоров и условий проведения процессов. Этот факт был установлен при исследовании продуктов гидротермальной обработки водных растворов «TiOSO4», «TiO(NO3)2» и H2[TiO(C2O4)2], а также аморфного геля гидроксида титана TiO2хH2O (синтезирован осаждением из раствора H2[TiCl6] избытком раствора аммиака). В процессе исследования варьировались следующие параметры: температура (в пределах от 150 до 250 С), продолжительность обработки (от 10 мин до– 6 ч) и концентрация исходного раствора прекурсора (в пределах 0,07 – 0,5 М).
Гидролиз «TiOSO4», «TiO(NO3)2», а также H2[TiO(C2O4)2] при всех исследованных концентрациях протекает полностью уже за 10 минут (как при 150, так и при 250 С). При этом образуются порошки TiO2 со структурой анатаза (размер кристаллитов 10 – 30 нм) (рис.15, 16). Увеличение продолжительности гидротермальной обработки до 6 часов выявило влияние этого фактора на структуру продуктов реакции.
Повышение температуры в и времени синтеза для систем, представляющие собой TiO2хH2O или раствор «TiOSO4» приводит лишь к росту размера кристаллитов анатаза (до ~ 35 нм), при этом образование TiO2 со структурой рутила не наблюдается. В то же время, образовавшийся в результате гидролиза «TiO(NO3)2» анатаз, при тех же условиях, в процессе его рекристаллизации практически полностью переходит в рутил (размер кристаллитов 65 – 70 нм). Из разбавленного раствора H2[TiO(C2O4)2] (0,07 М) при 150 С, не зависимо от времени термообработки, образуется смесь фаз (рутил и анатаз), а при температуры 250 С ( = 6 ч) образуется только фаза со структурой рутила.
Рис. 15. Дифрактограммы образцов TiO2, полученных высокотемпературным гидролизом 0,25 М раствора «TiO(NO3)2» при температуре 250 С в течение: а – 10 минут – анатаз, б – 6 часов – суперпозиция анатаза и рутила, в – 0,5 М раствора «TiO(NO3)2» при температуре 250 С в течение 6 часов – рутил
Рис. 16. Просвечивающая электронная микроскопия образцов TiO2, полученных высокотемпературным гидролизом «TiO(NO3)2»: а – 0,25 М раствора (Т =250 С, = 10 минут), б – 0,5 М раствора (Т = 250 С, = 6 часов), в – гидротермальной обработкой аморфного геля (250 С, = 6 часов)
3.3 Строение и свойства гидроксидов титана, циркония и олова (IV)
Гидроксиды титана, циркония и олова (IV) представляют собой полимерные оксо-гидроксо-ацидо-аквакомплексы, имеющие множество форм, которые грубо можно разделить на две основные: оловые (в которых атомы титана соединены между собой через группы ОН-) и оксоловые (атомы титана соединены через атомы кислорода). Однако образование конкретной формы возможно только при определённых, строго контролируемых условиях, и поэтому, реально формирующиеся в системе полимеры, зачастую, содержат в своём составе фрагменты обеих структур, а количественное соотношение этих фрагментов в рассматриваемых фазах зависит от условий получения последних. Кроме этого возможно образование и кристаллических форм ЭO2 при нейтрализации, например, сернокислых растворов Э(IV) вследствие локальных перегревов в системе за счёт экзотермичности данного процесса. При этом повышение температуры до 40 – 60С оказывается достаточной для образования устойчивых кристаллических зародышей указанных фаз. Этому также, вероятно, способствует структура комплексов, существующих в сернокислых растворах соединений Э(IV), т.е. соответствующая процессу кристаллизации ЭO2 структурная информация заложена в строении самих сульфатных комплексов.
В таблице 12 представлены данные по влиянию способа получения гидроксида титана (IV) на структуру продуктов его термического разложения в частности, состава растворов Ti (IV) и условий осаждения. В процессе термического гидролиза солянолянокислых и азотнокислых растворов соединений Ti(IV) осаждаются фазы переменного состава со структурой анатаза и рутила, тогда как из сернокислых растворов соединений Ti (IV) при тех же условиях выделяется только фаза со структурой анатаза.
Таблица 12.Результаты рентгенографического и деференциально – термического анализов гелей гидроксида титана (IV), полученных различными способами
|
№ обр. |
Способ синтеза гидроксида Ti (IV) |
[TiO2] % |
Структура |
Размер кристаллитов (нм) |
Температурные эффекты (С) |
|
|
Эндо- |
Экзо- |
|||||
|
1,2 |
H2[TiCl6] +NH3H2O н.у. |
71,7-78,0 |
Рентгеноаморфна |
- |
195-210 |
390-420 |
|
3 |
H2[TiCl6] + H2O кипячение |
87,4 |
Рутил (100%) |
7,9 |
200 |
- |
|
4 |
H2[TiCl6] + H2O кипяч.+2%зарод. |
80,9 |
Рутил (60%)+ анатаз (40%) |
9,2 |
210 |
- |
|
5 |
«TiOSO4» + NH3H2O н.у. |
71,7 |
Анатаз (100%) |
5,1 |
200 |
- |
|
6 |
«TiOSO4»+NaOH н.у. |
69,4 |
Анатаз (100%) |
4,6 |
210 |
- |
|
7 |
«TiOSO4»+H2SO4 +H2O, кип.+2%зар. |
82,2 |
Анатаз (100%) |
18,0 |
200 |
- |
|
8 |
«TiOSO4» +H2SO4 +H2O кипячение |
79,2 |
Анатаз (100%) |
15,6 |
190 |
- |
Согласно экспериментальным данным осаждение TiO2xH2O из разбавленных растворов соединений титана (IV) начинается при pH=1,4 (с.у.). Значения критического значения pH раствора, при котором начинается процесс осаждения геля, снижаются с ростом температуры раствора (до 0,47 при 100 С). Свежеосажденный при н.у. осадок хорошо растворяется в разбавленных минеральных и некоторых органических кислотах, легко пептизируется с образованием устойчивых коллоидных растворов при перемешивании с большим объемом воды. В щелочах растворимость незначительна: например, в 36 % - ном растворе NaOH она составляет 60 – 100 мг/л в пересчёте на TiO2.
При взаимодействии ацидокомплексов Ti (IV) с растворами щелочи (85 – 95 С) или металлического титана с азотной кислотой, а также при высокотемпературном гидролизе концентрированных растворов ацидокомплексов титана (IV) образуются -формы TiO2xH2O, не способные пептизироваться и практически не растворяющиеся в растворах щелочей и в разбавленных минеральных кислотах .
Гидроксид титана (IV), осажденный аммиаком из азотнокислых растворов соединений титана (IV), разлагается в интервале температур 20 – 350 оС, чему на дериватограмме соответствует размытый эндотермический эффект дегидратации (рис.16). Экзотермический эффект, связанный с кристаллизацией образующихся оксидов наблюдается при 404 – 476 С. Осадок TiO2xH2O, выдержанный при 200 С до постоянного веса, теряет до 17 масс. %, что соответствует ~ 1 молю воды на 1 моль TiO2.
TiO2xH2O, полученный методами осаждения или гидролиза, содержит переменное количество анионов и катионов, которые входили в состав прекурсоров. Если примесные катионы из системы можно практически количественно удалить из системы путём многократной промывки геля водой, то кислотные остатки SO42- и F-, связанные с основной фазой очень прочно и не удаляются при действии на продукт воды и разбавленных кислот.
При осаждении TiO2xH2O из солянокислого раствора H2[TiCl6] (0,01 – 1 моль/л) раствором аммиака, в его состав могут входить ионы Cl- и NH4+. В образцах, осаждённых при рН = 4 – 6, ион NH4+ не обнаружен, содержание же ионов Cl- увеличивается с ростом См раствора Н2[TiCl6], что объясняется снижением степени гидролиза указанной фазы при фиксированных условиях проведения процесса. Промывание этих осадков дистиллированной водой позволяет снизить содержание в них ионов Cl- в 1,9 – 2,8 раза, однако, полностью удалить ионы Cl- из системы не удаётся. Образцы же, осаждённые при рН > 7, содержат в своём составе переменное количество ионов NH4+, концентрация которых растет по мере уменьшения См исходного раствора Н2[TiCl6] и температуры синтеза. Содержание Cl- в этих осадках в 3,5 – 6,8 раз ниже, чем у образцов формировавшихся в кислых растворах и практически не изменяется в процессе обработки осадков водой.
Рис.16. ДТА гидроксида титана, осаждённого из азотнокислого раствора соединений Ti (IV).
Свежеосажденные при н.у. гидроксиды Ti(IV), согласно РФА, рентгеноаморфны, однако осадки, подвергшиеся длительному старению или полученные кипячением сульфатных растворов, обнаруживают слабые линии анатаза. В конечном итоге при длительном старении образуется продукт со структурой анатаза.
Исторически взгляды на строение гидроксидов Ti(IV) менялись несколько раз. Согласно начальным представлениям рентгеноаморфный TiO2xH2O представляет собой совокупность чрезвычайно мелких кристалликов TiO2, которые удерживают вокруг себя большое число адсорбированных молекул воды, т.е. фаза представляет собой просто «мокрый» кристаллический мелкодисперсный диоксид. При этом старение этой фазы рассматривалось как процесс низкотемпературной вторичной рекристаллизации, которая уменьшает суммарную поверхность порошка, а, следовательно, и число молекул воды, которое он может удерживать, что приводит к уменьшению реакционной способности фазы.
Другая точка зрения базируется на координационной теории, которая рассматривает явления, происходящие при гидролизе растворов комплексов Ti (IV). Согласно этим представлениям в процессе гидролиза наблюдается перенос протона от молекулы воды гидратированного катиона к ближайшей молекуле воды раствора. Моноядерные аквакомплексы Ti (IV) могут, по-видимому, существовать только в очень разбавленных растворах, так как они склонны к поликонденсации за счёт наличие в их составе полифункциональных групп О - Н. Гидролитическая полимеризация приводит к образованию оловых форм, в которых атомы титана связаны между собой мостиковыми OH – группами:
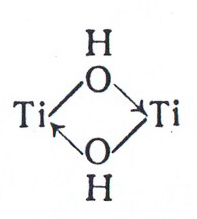
[ Ti (OH)(H2O)5]3+ [(H2O)4 (H2O)4 ]6+ + 2H2O
Дальнейшее протекание процесса связано с превращением оловых групп комплекса, отличающегося малой устойчивостью, в мостиковые оксогруппы:
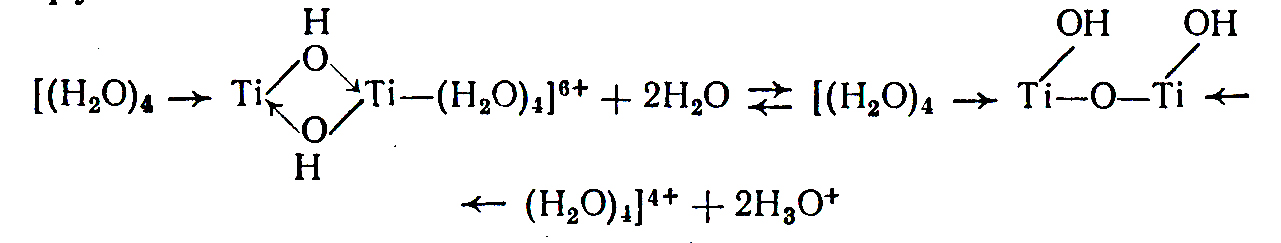
Экспериментально установлено, что при старении гелей гидроксида титана (IV) наблюдается уменьшение pH раствора, что подтверждается приведённоё схемой. Процессу оксоляции способствуют: а)повышение температуры системы, б) рост концентрации исходного раствора, в) использование термических, физико-химических или химических способов дегидратации. Обратный процесс (превращение оксоловых форм в оловые) самопроизвольно не протекает.
Гидролиз, оляция и оксоляция при их совместном протекании, приводят к образованию цепочек полиядерных комплексов, которые разрастаются до частиц коллоидных размеров:
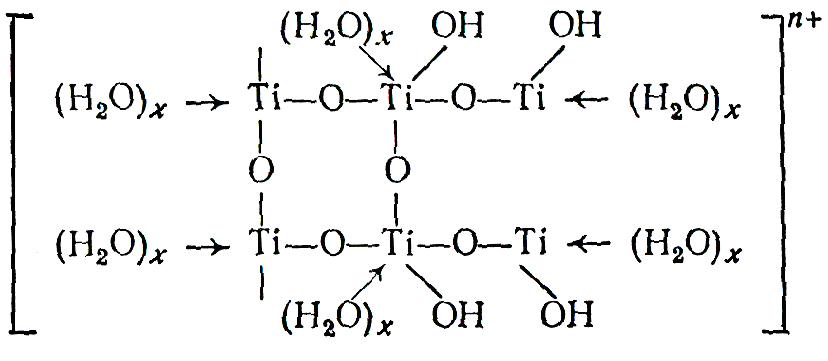
Помимо внутрисферной гидроксид обычно содержит значительное количество и внешнесферной воды, которая при нагревании постепенно отщепляется от полимера. Удаление же молекул воды из внутренней сферы комплекса вызывает его трансформацию в одну из форм кристаллического TiO2. Последний этап происходит в интервале температур 160 – 350 С.
ИК – спектр ксерогеля TiO2xH2O (форма, не содержащая внешнесферной воды) имеет три полосы поглощения с максимумами при 900, 678 и 535 см-1. Эти полосы можно отнести, соответственно, к колебаниям коротких концевых связей Ti – O, асимметричному валентному колебанию мостика Ti – O – Ti, соединяющего продольные ленты между собой в поперечном направлении, и валентным колебаниям фрагментов (OTi3) в лентах.
Дифрактограммы порошка гидроксида титана (IV) и плёнки HxTi2-x/4hx/4O4 толщиной меньше 10 нм подобны (рис. 17), что свидетельствует об их одинаковом слоевом строении. Единственный дифракционный пик на этих рентгенограммах соответствует межплоскостному расстоянию 6,75 для порошка и 10,00 для плёнки TiO2xH2O. При нагревании обоих форм TiO2xH2O до 200 С образуется анатаз с параметрами элементарной ячейки а = 3,77 и с = 9,51 .
Рис.17. Дифрактограммы: 1 - TiO2xH2O (порошок),
2 - TiO2xH2O (тонкая плёнка)
Тогда, согласно этим данным, химическую формулу гидроксида титана следует записать в виде H0.24Ti12O23(OH)2,2415,6H2O, структура этого вещества построена по типу H2V12O31nH2O и состоит из сдвоенных плоскостей TiO6 (межплоскостное расстояние 10,00 ± 0,05 ) между которыми находятся ионы гидроксония и молекулы воды (рис. 18).
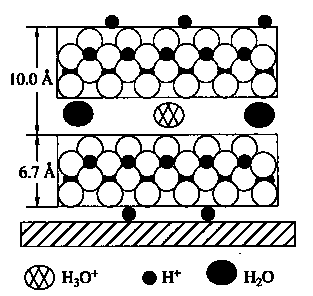
Рис.18. Модель структуры TiO2xH2O
Как отмечалось выше, гидроксид Zr (IV), в виде студенистого осадка, также как и гидроксид Ti (IV), можно получить действием щелочных растворов на водные растворы соединений циркония (IV). Фаза, полученная при н.у., в большей степени гидратирована и диспергирована, по сравнению с образцами, образующимися при нагревании. Аморфный гель гидроксида циркония (IV) в отличии от TiO2xH2O построен из тетрамерных комплексов [Zr4(OH)8(H2O)12](OH)8, соединённых между собой водородными связями. В частицу радиусом 2,4 нм входит примерно 70 таких тетрамеров, а условная её молярная масса составляет 5,410-4 г/моль при средней плотности порядка 1,55 г/см При нагревании золя до температуры его кипения диаметр рентгеноаморфных частиц ZrO2 ·xH2O увеличивается до 20– 64 нм.
Маловодный гидроксид циркония (IV) может быть получен путем гетерогенного взаимодействия между оксихлоридом или основным сульфатом циркония (IV) с водными растворами щелочи или аммиака. Состав этой формы представлен в таблице 18.
Таблица 18.Состав продуктов щелочного гидролиза некоторых соединений циркония (IV) при с.у..
|
Исходное соединение |
осадитель |
Состав продукта гидролиза |
||||
|
Me4+ масс.% |
OH-/Me4+ |
O2-/Me4+ |
Oок /Me4+ |
Фазовый состав |
||
|
Оксихлорид циркония |
10 % KOH |
54 |
1,5 |
0,6 |
0,7 |
0,7 «ZrO(OH)2»+ + 0,3 ZrO2 |
|
Основной сульфат циркония |
10 % KOH 10 % NH3·H2O |
43 38 |
2,0 2,3 |
1,0 0,9 |
- - |
ZrO(OH)2 0,3 ZrO0,5(OH)3 + + 0,7 ZrO(OH)2 |
В этом случае отдельные частицы гидроксида циркония имеют резко очерченные границы. Поверхность частиц сохраняется длительное время без видимых изменений в растворе и на воздухе, что в значительной мере отличает их от поведения частиц желатинообразных гелей гидроксидов, полученных осаждением из растворов ацидокомплексов.
При нагревании гидроксида циркония (IV), полученного любым из вышеописанных методов, согласно данным ДТА и ТГА наблюдается размытый эндоэффект отщепления воды при 100 – 350 оС, после чего убыль массы образца прекращается, а также экзоэффект при 410 – 530 оС (рис.19), связанный с процессом кристаллизации одной из полиморфных модификаций диоксида циркония. Интервал температур между окончанием эндоэффекта и началом экзоэффекта является температурной областью существования аморфной фазы при стандартном давлении.
В процессе низкотемпературной кристаллизации аморфной фазы образуется тетрагональная (t) модификация диоксида циркония, которая сохраняется при нагревании образцов до 900 оС. При более высокой температуре эта форма трансформируется в моноклинную (m) модификацию ZrO2. Процесс превращения t-фазы в m-фазу завершается при Т > 1080 оС (рис.20).
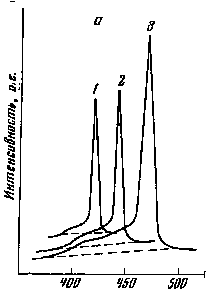
Рис. 19. ДТА кристаллизации при нагревании гидроксида циркония полученного: 1 – из тетрахлорида, 2 – из оксихлорида, 3 – из основного сульфата
Рис. 20. Дифрактограммы гидроксида циркония, полученного осаждением оксихлорида циркония раствором КOH (а) и продуктов его термическоко разложения при 390 С (б), 420 (в), 1000 С (г) и 1100 оС (д).
Нанокристалллы ZrO2, с достаточно узким распределением частиц по размеру, можно получить методом гидротермального синтеза. В частности, нанокристаллические порошки ZrO2 образуются при гидротермальной обработке водных растворов H2[Zr(NO3)6], а также аморфного геля «ZrO(OH)2». Синтез осуществляется в интервале температур 130 – 250 С и концентрации исходных растворов от 0,01 до 1,8 M (продолжительность гидротермальной обработки от 10 минут до 6 часов при давление насыщенного пара от нормального до ~ (4,0 – 5,0) ГПа).
Из водных растворов H2[Zr(NO3)6] методом высокотемпературного гидролиза, а также методом быстрого расширения сверхкритических растворов (RESS – методом), получают (m) модификацию ZrO2. Морфология и размер её кристаллитов (рис. 21) изменяются от квазисферических, диаметром 6 нм (RESS – метод), до анизотропных иглообразных - 7 - 26 нм (методом высокотемпературного гидролиза). С ростом температуры системы с 130 до 250 С размеры кристаллитов m – фазы ZrO2 увеличивается.
Таблица 14. Фазовый состав и физико – химические свойства нанокристаллических образцов диоксида циркония, синтезированных разными методами.
|
Методика |
Высокотемпературный гидролиз растворов H2[Zr(NO3)6] |
RESS |
Гидротермальная обработка “ZrO(NO3)2” |
||||||||
|
Концентра ция,моль/л Температу ра, К Время обработки Фазовый состав Средний размер кристаллитов (ПЭМ), нм Величина ОКР,±10%,нм Удельная поверхность БЭТ, м 2 /г |
1,8 523 6 ч 100% m 10Ч22 9 - |
0,25 403 1 ч 100% m 3Ч12 4 - |
0,25 403 6 ч 100% m 3Ч15 5 - |
0,25 523 1 ч 100% m 6Ч24 6 - |
0,25 523 6 ч 100% m 7Ч26 7 110 |
0,01 523 6 ч 100% m - 9 - |
0,38 523 4ГПа 0,5 ч 60%m +40%t - >60 - |
0,05 773 6 – 7 с 100% m 8 5 142 |
- 423 10 Мин 100% t 12 10 - |
- 523 1 ч 10%m +90%t 13 12 - |
- 523 6 ч 20%m +80%t 14 12 - |
Высокотемпературный гидролиз (Т =250 С) раствора H2[Zr(NO3)6] при Р = 4,0 ГПа, способствует образованию смеси m – и t – фаз ZrO2 и протекает практически полностью уже за 10 минут (как при 430, так и при 250 С). Скорость гидролиза H2[ZrCl6] ( в 0,25 М растворе) значительно меньше: при 150 С он завершается за 6 часов, при 250 С – за 1 час и приводит к образованию (m) модификации с размерами кристаллитов от 4 – 4,5 нм (150 С) и до 5 – 10 нм (250 С).
При гидротермальной обработке аморфного геля ZrO2·xH2O при130 С ( = 10 мин) образуется только t-модификация ZrO2 (размер кристаллитов от 10 до 12 нм), которая при повышении температуры до 250 С или времени синтеза до 6 часов постепенно превращается в m –фазу. Одновременно с этим происходит рост размеров кристаллитов и уменьшение величины удельной поверхности порошков.

Рис.21. Электронные микрофотографии (ПЭМ) и данные электронной дифракции нанокристаллических порошков ZrO2, синтезированных с использованием водных растворах H2[Zr(NO3)6]: а) – высокотемпературный гидролиз 0,25 М раствора (Т =250 С, = 6 часов), б – высокотемпературный гидролиз 0,25 М раствора (Т =130 С, = 1 час), в) – RESS – методом (Т =500 С, р = 100 МПа, = 6 секунд, 0,05М), г) – гидротермальной обработкой аморфного геля гидроксида циркония (Т =250 С, = 6 часов).
Возвращаясь к вопросу о строении гидроксида циркония (IV), необходимо отметить, что наиболее подробно в литературе рассматривается строение и свойства оксихлорида циркония, как типичного представителя ацидопроизводных этого элемента. Строение этой фазы было изучено методами рентгеноструктурного анализа, ПМР, КР и ИК спектроскопии (таб. 15).
Таблица 15. Состав частиц, обнаруженных в водных растворах оксихлорида циркония
|
Тип частицы |
Условия существования |
Метод исследования |
Литера-турный источник |
|
[Zr4+(OH2)n]4+ |
1-2 М HClO4, [Zr4+] = 0.02 М |
Спектрофо тометрия |
32 |
|
Zr3(OH)48+ |
2 М HClO4, [Zr] 510-4 М 1 М НСlO4 [Zr] 10-4 М |
||
|
Zr4(OH)88+ |
|||
|
Zr3(OH)57+ |
|||
|
Тример с зарядом +l |
1М (MeCl +HC1), где Ме = Li, Na и Cs |
Ультрацентри фугирование |
31 |
|
Zr[(OH)4Zr]n4+ n = 4-4.5 |
0.1 М НС1 + 1.9 М NaCl |
||
|
Zr[(OH)4Zr]n4+ n = 2-2.6 |
3М НС1 |
||
|
Тетрамер с зарядом zl |
0.05 М Zr4+ в 1 М НС1 или НСlO4 |
Ультрацентри фугирование |
33 |
|
Zr[(OH)4Zr]n4+ n < 2 |
5 М НС1 |
||
|
Zr[(OH)4Zr]n4+. n = 13 |
Осадок из H2SO4 |
Сорбция на катионите КУ-1 |
34 |
|
Zr[(OH)4Zr]n4+. n = 49 |
Осадок из HCl |
Для фазы состава ZrOCl28H2O, согласно РСА, характерно наличие тетрамерных гидроксокатионов [Zr4(ОH)8(Н2О)16]8+, существование которых, как было отмечено ранее, также зафиксировано в его концентрированных растворах, в которых они является доминирующими. Основываясь на полученных данных о пространственном расположении атомов Zr, О, С1, можно показать, что в оксихлориде циркония присутствуют три вида протонных группировок: мостиковые гидроксильные группы (две ОН-группы на один атом циркония) и два типа структурно неэквивалентных молекул воды. Четыре молекулы воды непосредственно координированы у иона циркония, а три - удерживаются во внешней сфере комплекса водородными связями. Согласно же результатам ИК спектроскопии, таких молекул воды, соответственно, две и пять, причем последние входят в комплекс по сэндвичеву механизму (также как в структуре гидроксида титана на рис.18), взаимодействуя с фрагментами Zr—С1 или Zr—ОН.
Установлено, что свежеосаждённый, тщательно отмытый водой гидроксид циркония содержит четыре гидроксогруппы приходящиеся на один атом циркония. При взаимодействии этой фазы с концентрированной соляной кислотой образуется «цирконил хлорид», строение которого рассмотрено выше. В данном случае в процессе образования формы принимают участие только две концевые гидроксогруппы, а две другие (оловые гидроксогруппы), более прочно связанные с центральным атомом, не замещаются на ионы хлора.
Анализируя обширный фактический материал о строении рассмотренных выше соединений циркония (IV), можно сделать вывод, что в этих фазах ни молекулы Н2О, ни группы ОН не являются строго индивидуализированными образованиями. Координационное число протона равно двум, вследствие чего он способен, формируя водородные связи различной прочности, входить в состав той или иной кислородно-водородной группировки. Действительно, при непосредственном исследовании методом ПМР гидратных оболочек ряда подобных соединений было обнаружено существование не только OH-групп и молекул воды, но и трехпротонных группировок. В то же время состояние гидратных оболочек соединений циркония может радикальным образом меняться в зависимости от изменения мольной доли воды в фазе, а также от вида и количества других ионов, сорбируемых фазой при обменных реакциях (в том числе в процессе соосаждения).
Исходя из тетрамерного строения комплексов циркония в солянокислом растворе, Whitney [42] предложил следующую схему образования и термического разложения гидроксида циркония (IV):
Zr4(OH)8(H20)l68+ + 8OН- [Zr4(OH)8(H20)16[OH]8]
[Zr4(OH)8(H20)16[OH]8] [Zr4(OH)8][OH]8 + 16Н2O
[Zr4(OH)8][OH]8 + 16Н2O [Zr4(ОН)2O6][ОН]2 + 6Н20
[Zr4(ОН)2O6][ОН]2 + 6Н20 4ZrO2 + 2Н2O
Очевидно, что эта схема отражает лишь идеализированный процесс. По-видимому, промежуточные фазы, характеризующие отдельные стадии представленной схемы, могут быть получены в определенных условиях, но реальный процесс, несомненно, сложнее.
В настоящее время считается, что первичный золь гидроксида циркония (IV) представляет собой совокупность полимерных катионных форм, в основе которых лежит циклический комплекс состава [Zr4(OH)8(H2O)l6]8+.
Наличие этого цикла характерно и для многих других соединений циркония (IV), например, он обнаружен в структурах оксигалогенидых, оксалатных, карбонатных, фторидных, сульфатных, азотнокислых и других фазах, являющихся продуктами гидролиза основных форм. С изменением концентрации раствора и его рН, эти комплексы могут взаимодействовать между собой с образованием более крупных агрегатов. При этом главная роль в формировании таких форм принадлежит ол-связям1, которые образуются между атомами циркония, благодаря обобщению гидроксогрупп. Полимеризация становится необратимой при переходе ол- в оксо-связи2, в частности при нагревании, что ведет к формированию частиц диоксида циркония. Процесс оксоляции свежеосажденных гидроксидов циркония заканчивается, в основном, при выдерживании их на воздухе до постоянной массы. Дальнейшая интенсификация процесса старения гидроксида циркония возможна за счет увеличения температуры маточного раствора выше его точки кипения, т.е. в гидротермальных условиях.
Рентгенофазовый анализ образцов указывает на постепенный характер процесса перехода частиц из аморфного состояния в кристаллическое примерно после 24 ч термообработки (кипячение раствора). С увеличением времени термообработки растёт число кристаллических частиц в системе и их размер. В аморфном гидроксиде (-фаза) атомы циркония соединены между собой через двойные оловые мостики, которые в дальнейшем превращаются в оксо-связи (-фаза) (рис.22). Если гидроксиду приписать формулу Zr(ОН)4nН2O, то продукты реакций оксоляции и дегидратации оксогидраты можно выразить в виде: ZrOx(OH)4-xnН2О. -фаза может рассматриваться как совокупность тетрамерных комплексов Zr4(OH)16(H2O)8, которые склонны к объединению через мостиковые группы О - Н, что приводит к образованию геля гидроксида циркония (IV).
Установлено, что с повышением рН растворов скорости процессов старения гидроксидов и кристаллизации аморфных осадков увеличиваются [48-53]. Механизм каталитического действия гидроксил-ионов в этих процессах заключается в замещении молекул воды в координационной сфере комплексов, что приводит к росту скорости реакции оксоляции.
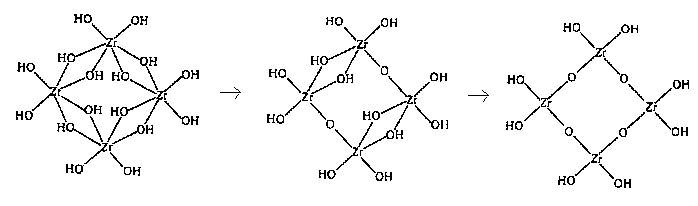
– – –
Рис. 22. Строение -, -, -гидроксида циркония
Это связано с тем, что процесс оксоляции протекает в результате взаимодействия двух гидроксильных групп, поэтому введение вместо молекул воды в координационную сферу комплексов дополнительных гидроксогрупп из раствора, пространственно облегчает нахождение группы – партнера, принадлежащей другому центральному атому. Реакция оксоляции практически необратима, поэтому и при большой концентрации ОН-групп в системе, процесс быстро заканчивается образованием оксосвязей.
Гидролиз соединений циркония (IV), также как соединений титана (IV), начинается при низком значении pH раствора и в значительной степени зависит от его концентрации, температуры и наличия в системе посторонних фаз. В солянокислых растворах при Т~5 оС осаждение гидроксида начинается при pH = 1,9, в сернокислых растворах при pH = 1,7. В обоих случаях осаждение заканчивается при pH = 4,2.
Литературные данные о природе так называемых «оловянных кислот» крайне противоречивы. Одни исследователи считают их гидратами определенного химического состава, другие – гидратированными оксидами, где термин «гидратированный» означает отсутствие стехиометрически связанной воды в фазе. На основании данных рентгенофазового анализа (РФА) и метода парамагнитного резонанса (ПМР) ряд исследователей определяют «оловянные кислоты» как агломераты высокодисперсных частиц диоксида олова, поверхность которых покрыта сеткой гидроксильных групп и связанных с ними молекул воды. По спектрам широкополосного ПМР численным интегрированием экспериментально наблюдаемой первой производной линии поглощения, рассчитаны величины вторых моментов М220°С для образцов SnO2xH2O, предварительно отоженных при разных температурах.
На рис.23 представлен общий ход зависимости М220°С=F(T), подтверждающих кластерную модель для состояния воды в данных гидроксидах. Действительно, спектры ПМР образцов, отожженных при Т»200°С, характеризуются величинами М220°С, обычными для малоподвижных (структурных), изолированных ОН-групп. Присутствие в неотоженных образцах рядом с такими ОН-группами подвижных молекул сорбционной воды приводит к усреднению локальных полей, проявляющемуся в сильном сужении линий резонансного поглощения.
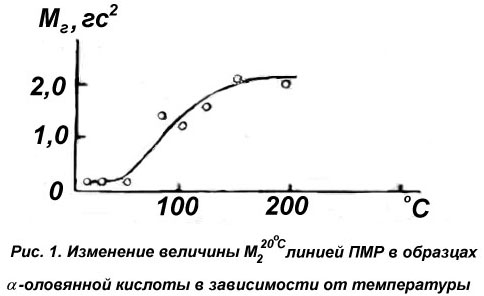
Рис.2 Изменение величины М220°С линией ПМР в образцах
a- SnO2xH2O в зависимости от температуры
Отжиг при сравнительно низких температурах сопровождается, в основном, потерей наименее связанной адсорбционной воды. В результате, растет масса кластеров «бесподвижной воды», что и увеличивает М220°С. Заметим, однако, что применение ПМР, показавшее неоднородное распределение протонов в SnO2xH2O само по себе не говорит о том, что эти фазы и продукты их частичной дегидратации являются определенными соединениями.
Изучение «оловянных кислот» с помощью эффекта Мессбауэра и РФА, было показано, что они являются высокодисперсными системами {SnO2-H2O}, в которых гранулы геля характеризуются средним диаметром » 20. Такие размеры частиц SnO2 предполагают значительную долю атомов олова, находящихся на их поверхности. Так как координация атома на свободной поверхности ниже, чем координация внутреннего атома, следовало бы ожидать сложного спектра мессбауэровского поглощения исследованными образцами. Однако экспериментально этого не наблюдалось. Напротив, резонансные линии всегда имели нулевой химический сдвиг относительно источника SnO2, что говорит о том, что квадраты волновых функций около ядер олова в «оловянных кислотах» и в безводном SnO2 не изменяются. Отсутствие во всех случаях заметных изменений формы и ширины резонансной линии также говорит о том, что ближайшее окружение атомов олова не меняется в ходе превращения гидроксидов и соответствует октаэдру из атомов кислорода или других кислородсодержащих лигандов.
Это обстоятельство заставляет предположить, что атомы олова на поверхности микрокристаллов SnO2 повышают свою координацию до координации внутренних атомов. Было бы естественно допустить, что необходимый для этого кислород поверхностные атомы олова получают присоединением ОН-групп, или молекул воды. Однако, даже в последнем случае, включение атома кислорода в ближайшую координационную сферу поверхностного атома олова может привести к изменению первоначального расстояния между протонами за счет смещения одного из них в сторону ближайшего атома кислорода, что равносильно образованию двух ОН-групп.
Изложенные выше выводы одинаково хорошо подтверждается данными ПМР и эффекта Мессбауэра. Такой подход позволяет также понять различие результатов экспериментов по дифракции электронов и рентгеновских лучей. Действительно, высокопроникающее рентгеновское излучение должно в основном давать информацию о порядке внутри кластеров SnO2; напротив, электронные дифрактограммы характеризуют лишь структуру их поверхностных слоев.
Представленные данные позволяют понять безуспешность попыток составления эмпирических формул, связывающих общее содержание воды в «оловянных кислотах» с общим содержанием SnO2. Однако не менее очевидно то, что «оловянные кислоты» не просто «мокрый SnO2», что подтверждается высокой сорбционной активностью гидратированного диоксида Sn(IV), связанной с наличием в системе поверхностных функциональных групп ОН– и молекул Н2О (рис.24).
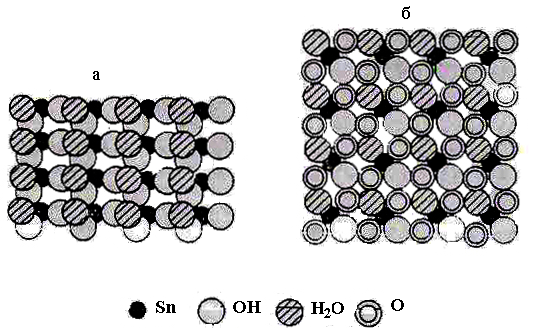
Рис.24. Поверхностная структура гидратированного диоксида олова.
Расположение молекул воды и ОН- -групп: а)- на плоскости (100); б)- на плоскости (001).
Необходимо отметить, что прочность поверхностных связей
Ме-ОН зависит от количества координированной ионом олова молекул воды. Следовательно, прочность указанных связей и их кислотные свойства на разных кристаллографических плоскостях будут различными. В соответствии с предложенной моделью наиболее кислотными свойствами обладают поверхностные ОН--группы, расположенные на плоскости (001), и количественное соотношение трех типов ОН--групп в порядке убывания кислотной силы равно 1:2:2.
Помимо чисто поверхностных гидроксильных групп могут быть и, так называемые, внутриглобульные, которые были обнаружены в некоторых образцах, подвергшихся гидротермальной обработке. Впрочем, хемосорбция воды, объясняемая возникновением водородных и донорно-акцепторных связей, не обязательно завершается образованием гидроксильных групп. По-видимому, состояние протонов в связанной воде в общем случае таково, что даже при твердо установленном брутто-составе гидрата, эти протоны нельзя всецело относить ни к гидроксильным группам, ни к структурным аквогруппам. Более отдаленные слои сорбированной воды в рассматриваемых фазах также структурированы и связаны с первичным гидроксильным покровом силами диполь-дипольного взаимодействия.
Таким образом в системе (SnO2-H2O) вода может быть связана различно: химически, осмотически, капиллярно, и эти типы оксигидратов могут находиться в состоянии истинного или псевдоравновесия. Поэтому, при непрерывном обезвоживании подобных препаратов трудно ожидать, что на определенных этапах будут формироваться фазы определенного состава: кривая обезвоживания в этом случае монотонная и лишена характерных ступенек.
При термообработке гидратированного диоксида олова, полученного золь-гель методом, также наблюдается непрерывная дегидратация, под которой понимают: удаление воды, связанной с поверхностью; дегидроксидирование поверхности, которое обычно изображают как процесс удаления концевых гидроксидных групп:
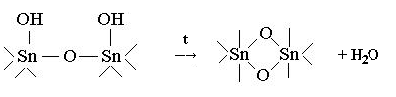
и протекание оксоляционно-химических процессов на границах между блоками и на дефектах структуры, которые представляют собой превращение диоловых связей (мостиковых ОН-групп) в оксосвязь:
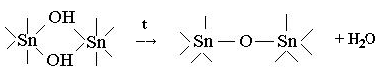
Дегидратация материала практически заканчивается при 500°С, однако небольшое количество воды сохраняется при более высокой температуре: в частности, с использованием ИК и КР спектроскопии, в кристаллическом диоксиде олова обнаружены октаэдрические гидроксостаннатные группировки [SnOx(OH)6-x](2+x)-, где х может иметь значения от 0 до 6. На рисунке 25 приведены кривые ДТА и ТГА для образца гидратированной двуокиси олова, полученной золь-гель методом.
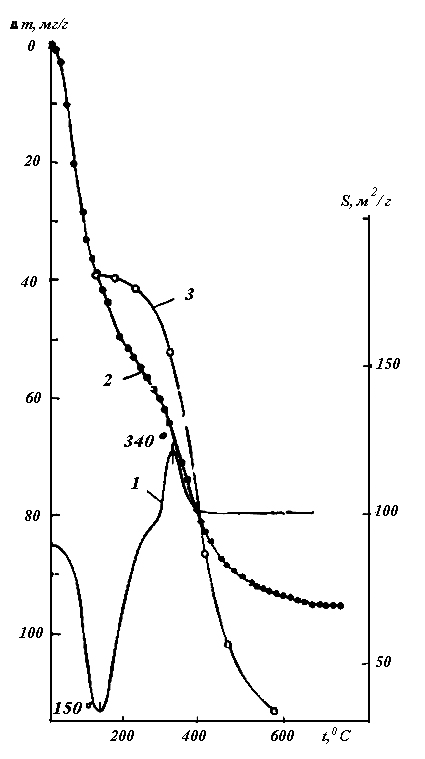
Рис. 25. Кривые ДТА и ТГА образца гидратированного диоксида олова (ГДО), синтезированного золь-гель методом (1-кривая ДТА, 2- кривая ТГА, 3- сокращение удельной поверхности в процессе ДТА)
Химические превращения в гидратированном диоксиде олова при его термообработке можно представить схемой:
-xH2O -(2-x)H2O
{SnO2}Sn(OH)4 ® {SnO2}SnOx(OH)4-2x ® SnO2
Использование метода комбинационного рассеяния света позволило обнаружить в продуктах гидролиза SnCl4 и наличие группировок
[Sn(OH)6-xClx]2-. С уменьшением атомного отношения Cl/Sn наблюдается превращение оловогалогенных группировок в гидроксостаннатные и оксидные. Поверхностный слой можно условно изобразить в виде:
{SnO2}SnOx(OH)yCl4-2x-y, где 0 < x < 2,0 < y < 4.
При осаждении SnO2xH2O из растворов Н2[SnCl6] с помощью NH3H2O образуется гель, содержащий по данным РФА кристаллический NH4Cl, удерживаемый сетчатой структурой геля.
Если процесс гидролиза соединений олова (IV) протекает при низких температурах, то образующаяся форма a-SnO2·хH2O, легко растворяющийся в кислотах и в меньшей степени в щелочах:
a- SnO2·хH2O + KOH = K2[Sn(OH)6],
a- SnO2·хH2O + HNO3 = H2[Sn(NO3)6] + H2O.
В то же время гидрат диоксида олова, осажденный при высокой температуре или образовавшийся при растворении олова в азотной кислоте (b-форма), весьма инертен. При хранении на воздухе или при нагревании
a-форма гидроксида «стареет», превращаясь в b-форму, которая отличается большей химической инертностью. В обоих случаях получается рыхлый белый осадок, который при высушивании постепенно теряет воду и превращается в твердый порошок. При прокаливании вода также постепенно удаляется и остается чистый диоксид олова.
Таким образом, анализ представленных данных показывает, что SnO2xH2O, осаждённый при низких температурах из растворов ацидокомплексов представляют собой полимерные оксо-гидроксо-аквакомплексы оловой структуры, состав которых зависит от условий синтеза фазы. Так же как в случае гидроксида титана -SnO2xH2O, при старении постепенно трансформируется сначала в оксоловую форму, а затем образуется оксид олова (IV). При этом, за счёт более высокого поляризующего действия катиона Sn4+, по сравнению с аналогичными ионами титана и циркония, стабильность гидратных форм в случае олова ниже, по сравнению с аналогичными формами титана и циркония.
3.4. Ионообменные свойства ЭO2xH2O
В рассмотренных гидроксидах ЭO2xH2O оловые и концевые гидроксильные группы, а также молекулы воды, могут замещаться различными анионами. Степень замещения зависит от относительной концентрации реагентов и способности аниона координироваться с атомом центрального элемента, а также от продолжительности предварительного старения. В кислых растворах рассматриваемые фазы проявляют анионобменные свойства, а в щелочных – катионобменные (акво- и гидроксогруппы диссоциируют, отщепляя протон) т.е. ЭO2xH2O функционирует как амфотерный гидроксид.
Величины и pK гидроксо-групп для TiO2xH2O определяли методом потенциометрического титрования раствором LiOH при стабилизирующей ионной силе раствора (I = 0,1), достигаемой с помощью LiCl. На кривой потенциометрического титрования TiO2xH2O (рис.26) можно выделить три участка с pK1 = 5,0 ± 1,0, pK2 = ± 7,9 ± 1,7 и pK2 = 10,6 ± 0,4.
Рис.26. Кривая потенциометрического титрования TiO2xH2O
При динамическом режиме сорбции ионов щелочных металлов из растворов с pH > 12,2, для TiO2xH2O получены следующие значения обменной емкости (): Li+ - 0,48, Na+ - 0,41, K+ - 0,34, Rb+ - 0,32 и Cs+ - 0,31 мг-экв/мг-моль TiO2. Понижение по ряду ионов Li+ > Na+ > K+ > Rb+ > Cs+ связано с канальной структурой ионообменника (рис.18, табл.16). Значительная сорбционная способность гидроксидов титана (IV) связана те только с высокой концентрацией у них сорбционных центров, но и большой удельной поверхностью образцов. Так как и первый и второй факторы, предопределяющие сорбционную ёмкость различных форм этих гидроксидов, зависят от способов их получения, данные о величине () для них по отношению к различным катионам и анионам противоречивы.
Таблица 16. Зависимость сорбционной ёмкости от размера сорбируемого катиона.
|
Ион |
D1, |
, мг-экв/мг-моль |
||
|
Свежеприготовленный TiO2xH2O |
Воздушно-сухой |
Высушенный при 105 С |
||
|
Li+ |
1,56 |
0,48 |
0,46 |
0,39 |
|
Na+ |
1,96 |
0,41 |
0,30 |
0,19 |
|
K+ |
2,60 |
0,34 |
0,27 |
0,18 |
|
Cs+ |
3,30 |
0,31 |
0,22 |
0,10 |
|
N(CH3)4+ |
4,50 |
0,28 |
0,14 |
0,10 |
|
N(C2H5)4+ |
7,00 |
0,06 |
- |
0,03 |
Известно, что при нагревании TiO2xH2O происходит обезвоживание данной фазы за счёт конденсации OH – групп, сопровождающееся уменьшением в ней соотношения OH- : Ti4+, что ведет к снижению значений (). В большей степени это относится к сорбенту в H – форме. Сорбент же в Li – форме характеризуется более высокой термической устойчивоcтью (рис. 27) и после нагревания до 480 – 520 С приобретает селективность по отношению к ионам Li+.
Взаимодействие аморфного, маловодного гидроксида титана, полученного с использованием в качестве прекурсора «титанилсульфата» аммония, с водными растворами нитрата и ацетата свинца показывает, что степень сорбции катиона в данном случае зависит от природы аниона, присутствующего в системе. Установлено, что при температуре 20 и 90 С высокая скорость сорбции ионов свинца твердой фазой наблюдается в первые 2 – 3 часа (рис.28), а равновесие между жидкой и твердой фазами устанавливается через 5 – 10 часов.
Рис. 27. Дериватограммы TiO2xH2O в Н-форме (а) и Li-форме (б)
На начальном этапе взаимодействия (1 – 2 ч) повышение температуры системы с 20 до 90С существенно увеличивает скорости процесса. При взаимодействии гидроксидов Ti(IV) с раствором нитрата свинца содержание ионов свинца в сорбенте по окончании процесса не превышает 30 моль %, а при замене нитрата на ацетат свинца той же концентрации () сорбента по ионам Pb2+ возрастает практически в два раза (рис.28).
Изменение () сорбента при переходе от нитратных к ацетатным растворам солей свинца может объясняться несколькими факторами: а) более высокой () гидроксида титана по отношению к ацетат – ионам, по сравнению с нитрат – ионам; б) более высокой степенью гидролиза ионов свинца в растворе его ацетата. Первый фактор вызывает необходимость перехода (для сохранения электронейтральности сорбента) дополнительного числа ионов Pb2+ в твёрдую фазу, а второй - предопределяет высокую концентрацию в растворе ионов Pb(ОН)+, переход которых в сорбент (по мнению авторов исследования) предпочтительнее, чем ионов Pb2+.
Рис.28. Изменение () гидроксида титана во времени при сорбции ионов свинца из 0,5 моль/л растворов: (1, 1”) азотнокислого свинца и (2, 2”)ацетата свинца.( 1,2 - 20 С; 1”, 2” – 90 С.)
Отметим, что если роль первого фактора не вызывает сомнения, то заявленное влияние степени гидролиза катиона на () гидроксида титана не может быть признано удовлетворительным. Возражения связаны с тем, что авторами не учитывается: а) снижение () сорбентов рассматриваемого типа по мере роста радиуса сорбируемого катиона и уменьшение его заряда; б) влияние рН растворов на константу равновесия, описывающую рассматриваемый процесс. В связи с этим отметим работу [17], авторами которой исследованы зависимости () гелеобразных форм TiO2xH2O от рН раствора сорбата и времени сорбции (рис.29 и 30). Необходимое значение рН (2 – 6) раствора Pb(NO3)2 достигалось за счёт введение в него, на этапе приготовления, 2% растворов HNO3 или NH Степень сорбции (соотношение СМ Pb2+/CМ /Ti4+) определяли по остаточному значению СМ Pb2+ в жидкой фазе. Из представленных данных (рис.29) следует, что () гелеобразных форм TiO2xH2O по отношению к ионам свинца увеличивается практически в два раза при изменении рН сорбата от 4 до 6, т.е. концентрация ионов гидроксония в системе играет значительную роль в смещении равновесия процесса сорбции. рН 0,5М раствора Pb(NO3)2 при с.у. равен 4,3, а рН раствора ацетата свинца той же концентрации составляет 5,5. В связи с этим можно утверждать, что значительную роль в повышении (), при переходе от первой системы ко второй, играет снижение концентрации ионов Н3О+ в растворе сорбата (при замене нитрата свинца на его ацетат).
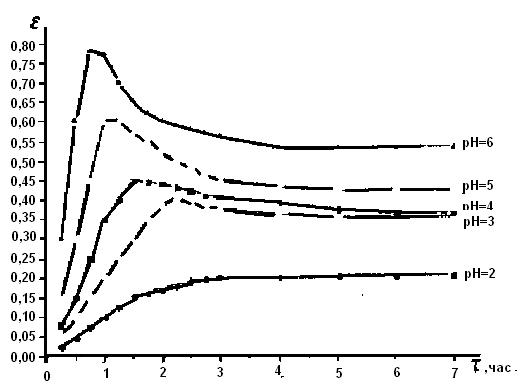
Рис.29 . Зависимость степени сорбции СМ Pb2+ / CМ Ti4+ от времени () при различных значениях рН раствора сорбата (исходная концентрация раствора свинца II равна 0.1 М)
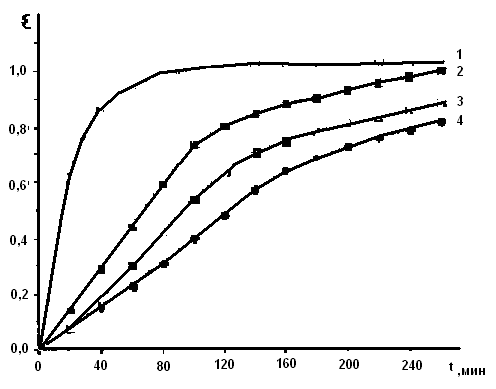
Рис.30. Зависимость степени сорбции СМ Pb2+ / CМ Ti4+ для гелей, полученных из азотнокислых растворов Ti (IV): 1 – 0,01 М, 2 – 0,05 М, 3 – 0,25 М, 4 – 1 М, от времени сорбции ()
Этим же объясняется и аномальное изменение () рассматриваемых фаз во времени: в процессе сорбции происходит замещение ионов водорода в функциональных группах на катионы свинца, что снижает рН раствора сорбата и способствует росту скорости десорбции ионов Pb2+.
Аналогичные результаты получены и при сорбции ионов свинца гидроксидом циркония (рис. 32).
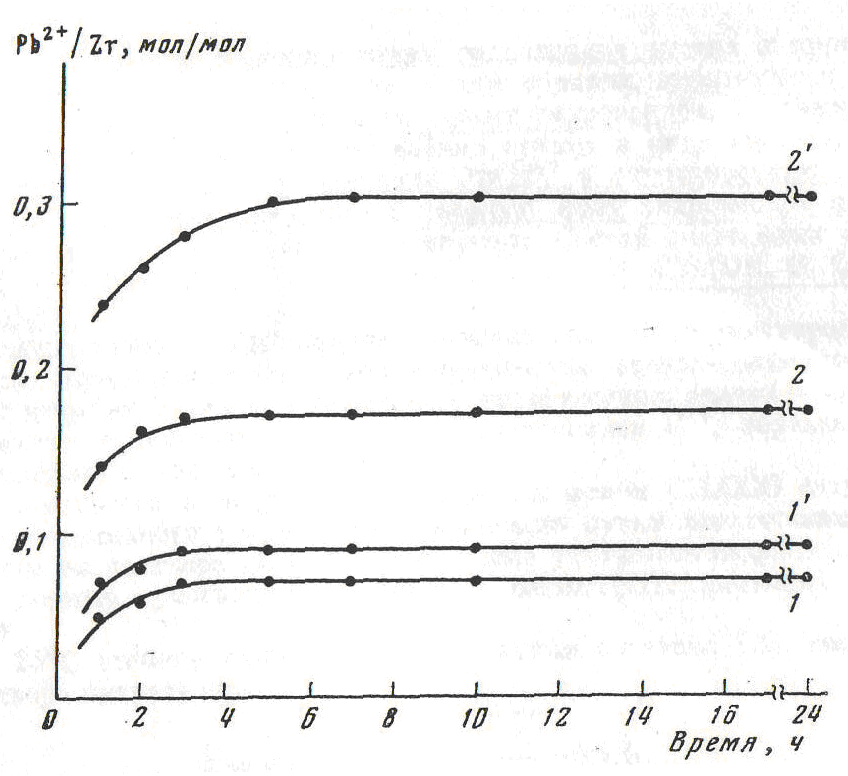
Рис.32. Поглощение свинца гидроксидом циркония из 0,5 моль/л растворов азотнокислого свинца (1, 1”) и ацетата свинца (2,2”).1,2 - 20 С; 1”,2” – 90 С.
Равновесие между жидкой и твердой фазами устанавливается за 3 –5 часа, в зависимости от условий изготовления образцов. На начальном этапе взаимодействия (1 – 2 ч) повышение температуры системы с 20 до 90 оС существенно увеличивает скорость реакции. Продукты взаимодействия гидроксида циркония с растворами солей свинца рентгеноаморфны. При их нагревании вначале наблюдается образование диоксида циркония тетрагональной или кубической модификации, а при более высоких температурах (выше 700 оС) в системе формируется моноклинная форма ZrO2 и PbZrO
Сорбционная обменная емкость гидроксидов рассматриваемого типа существенным образом зависит от состава и концентрации контактирующего раствора. Характер процесса меняется от анионо- к катионнообменному при переходе через изоэлектрическую точку. Зависимости СОЕ (сорбционной обменной ёмкости) от величины pH для образцов гранулированного гидроксида циркония представлены на рисунке 31, полученных при комнатной температуре - (образец I) или отожжённых при 300 С в течении 20 мин, (образец II).
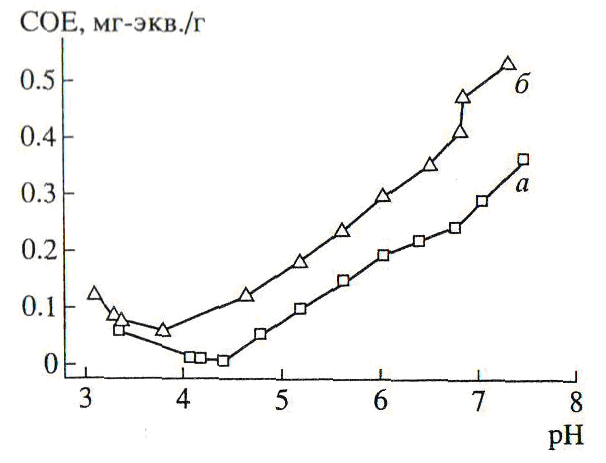
Рис. 31. Поглощение ионов свинца гидроксидом циркония из 0,6 моль/л азотнокислого раствора в зависимости от его рН: (а) – образец I, (б) – образец II.
Следует отметить, что термообработка ZrO2·xH2O приводит к увеличению СОЕ, а следовательно, и росту числа доступных обменных центров.
Значения показателей констант, характеризующих равновесия на границе раздела гидроксид / электролит (pK), а также значения pHт.н.з (т.н.з. – точки нулевого заряда) представлены в таблице 17. В других работах, например в [55], значения pK1 и pK2 для гидроксида циркония равны соответственно 8 и 10,5, что указывает на зависимость этой величины от способа получения гидроксидов.
Таблица 17. Значения показателей констант (pK ± 0,1) кислотно – основных равновесий на границе ZrO2xH2O / / электролит
|
pKi0 |
Высушивание при 50 оС |
Высушивание при 120 оС |
Высушивание при 200 оС |
|
pK10 pK20 pK30 pK40 pHт.н.з |
4,5 8,8 6,3 7,1 6,8 |
4,7 8,9 6,3 7,1 6,6 |
4,9 9,2 6,7 7,1 7,0 |
Кислотные и особенно основные свойства гидроксида циркония зависят от числа активных центров, способных проявлять кислотные или основные свойства, т.е. от количества концевых OH – групп в отдельных частицах.
Образцы сорбентов, осаждённые из кислых растворов ацидокомплексов Zr(IV) представляют собой фазы переменного состава, например: MeO2(H2O)х(OH)y(NH4)z(X)k, где X – Cl , NO3, CH3COO и (y + k) = z MeO2(H2O)х(OH)y(NH4)z(SO4)k/2. Взаимодействие этих фаз с растворами солей щелочноземельных элементов и свинца осуществляется в форме количественного обмена между Men+ и NH4+, одновременно с этим наблюдается частичный обмен Me2+ <--> 2H+, что приводит к снижению pH раствора сорбата. Значения сорбционной ёмкости образцов () гидроксидов Zr (IV) зависит от условий осаждения данных фаз: она уменьшается по мере увеличения концентрации исходных растворов соединений циркония, из которых сорбенты были осаждены, а также с ростом температуры осаждения указанных фаз. При фиксированных параметрах синтеза (концентрация, температура) максимальное значение наблюдается у форм, осаждённых из нитратных растворов соединений Zr (IV). На примере процессов сорбции ионов Pb2+ , для этих форм было установлено:
а) max увеличивается с ростом pH исходного раствора Pb(NO3)2; б) зависимость () имеет экстремальный характер при pH = 4 – 6; в) с ростом концентрации сорбируемых ионов в сорбенте, pH раствора соли снижается, что способствует протеканию процесса десорбции, так как процесс сорбции между MeO2xH2O и растворами солей носит обменный характер.
Равновесие в системе наступает через 2 – 3 часа после начала процесса. Также установлено, что значение для – ZrO2xH2O на 12 – 18 % ниже, чем у – TiO2xH2O и в большей степени зависит от значения pH раствора Pb(NO3)2.
Ионообменные свойства гидроксид циркония проявляет и по отношению к ионам Cu2+, Co2+, Ni2+, при этом его свойства так же зависят от способов получения и условий предварительной обработки. В частности, наибольшей величиной СОЕ обладает образец, осажденный раствором NaOH, немного меньшую величину СОЕ имеет образец, осажденный раствором KOH, и почти в 2 раза меньшей емкостью обладает образец гидроксида циркония, осажденный раствором аммиака. Представленные данные показывают, что на сорбционные свойства гидроксида циркония оказывают влияние следующие условия его синтеза:
а) природа катиона, входящего в состав осадителя (образец, осажденный с помощью раствора NaOH, имеет более высокое значение СОЕ по сравнению с образцом, осажденным раствором аммиака);
б) увеличение времени созревания первичного геля ведет к снижению содержания OH – групп, т.е. к снижению значения СОЕ;
в) увеличение температура сушки приводит к снижению СОЕ, за счёт уменьшения его активной поверхности.
Исследование сорбции ионов Cu2+, Co2+, Ni2+ гидроксидом циркония показало, что по СОЕ исследуемые ионы располагаются в ряд: Cu2+ > Co2+ > Ni2+, т.е СОЕ снижается при увеличении кристаллографического радиуса катиона.
В процессе реакции между ZrO2xH2O и Mе(OH)2 (где Ме – ионы щелочноземельных элементов) образуются как аморфные, так и кристаллические фазы. Состав аморфных фаз переменный: максимальное соотношение M2+/M4+ в синтезированных продуктах составляет: Ba2+/Zr4+ = 1,35; Sr2+/Zr4+ = 0,9. Превращение аморфных фаз в кристаллические соединения ускоряется при повышении температуры и концентрации раствора M(OH)2.
Таким образом, можно сделать вывод, что различные формы гидроксида циркония (IV) имеют различное строение, зависящее от способов синтеза фазы, а, следовательно, и различные свойства. Наиболее активны их оловые формы, синтезированные при низких температурах и представляющие собой ацидогидроксоаквокомплексы переменного состава
Гидрат диоксида олова сорбирует из раствора многие содержащиеся в нем частицы. В кислых средах SnO2xH2O обладает анионообменными свойствами, при этом гидроксил-ионы замещаются на эквивалентное количество анионов раствора. С повышением рН раствора проявляются катиоообменные свойства гидроксида олова (IV). По своим сорбционным свойства эти фазы не уступают аналогичным формам гидроксидов титана и циркония, а в ряде случаев превосходят последние. Так золи SnO2xH2O адсорбируют из концентрированных растворов перекиси водорода (84-87 масс.%) ионы Al(OH)2+, Fe(OH)2+, CuOH+ и др. (Этот эффект используют для предохранения перекиси водорода от каталитического разложения).
3.5. Неорганические полимеры.
Неорганические полимеры делятся на гомоатомные и гетероатомные. Среди неорганических полимеров гомоатомные встречаются относительно редко. К ним, в частности, относятся полимерные формы серы, селена, теллура, фосфора, мышьяка, сурьмы, висмута и некоторых других элементов, а также кремневодороды и сульфаны.
Как известно, наиболее разнообразные гомоцепи образуют атомы углерода. Это связано с тем что, у атомов углерода число валентных орбиталей равно числу валентных электронов, что, с учётом небольшого атомного радиуса этого элемента, даёт возможность образовывать прочные цепи, характеризующиеся наличием между атомами, как одинарных, так и кратных связей. В то же время соседние атомы бора и азота образуют гомоцепи, энергия связи между атомами в которых значительно ниже, чем в гомоцепях, образованных атомами углерода. В первом случае это связано с бльшим, чем у атома углерода атомным радиусом и меньшим числом валентных электронов, а во втором - с наличием у атома азота неподелённой электронной пары, которая дестабилизирует связь N – N. Очевидно, что сочетание атомов бора и азота в одной системе нивелирует недостатки каждого из них, т.к. число валентных электронов в такой системе будет равно числу валентных орбиталей. В результате этого образуются гетероатомные аналоги соединений и простых веществ углерода: (BN)n – слоистый полимер (аналог графита), (BN)n, в котором орбитали и атома бора, и атома азота находятся в состоянии sp3- гибридизации (аналог алмаза), циклический аналог бензола - B3N3H6 и бутадиена – B2N2H6 (рис.33 и 34), а также каучука (- B2N2H4 -)n. Ряд подобных фаз предлагаем продолжить читателям. Аналогичные формы полимеров возникают и при использовании пар атомов В – Р и Р – N. При этом, с учётом большого числа орбиталей у атома фосфора (свободный d-подуровень) стабилизировать данные формы можно за счёт введения в систему периферийных донорных атомов. 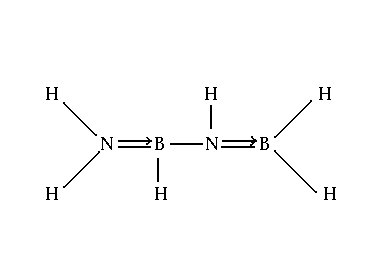
Рис. 34. Гетероатомный аналог бутадиена –1,3
Рис.3 Гетероатомные аналоги – сверху вниз – бензола, дифенила и нафталина.
.
Рассмотрим состав и строение гетероатомных полимеров, на основе цепей состоящих из атомов фосфора и азота (рис.35). Например, фосфонитрилхлорид образуется при взаимодействии хлорида фосфора с хлоридом аммония:
nPCl5 + nNH4Cl = (PNCl2)n + 4nHCl
Наличие в полученной системе атомов хлора приводит к образованию электронизбыточного соединения (6 электронных пар атомов хлора на один атом фосфора, у которого после образования связей с азотом остаётся только три свободные орбитали). Тогда для стабилизации формы необходимо заменить связи P – Cl на связи Р – О, Р – S или P – N: [PN(OR)2]n, [PN(SR)2]n, [PN(NHR)2]n. Последние фазы называются фосфазенами. Они в отличие от фосфонитрилхлорида устойчивы к действию воды и, в целом, достаточно химически инертны.
 а)
а)
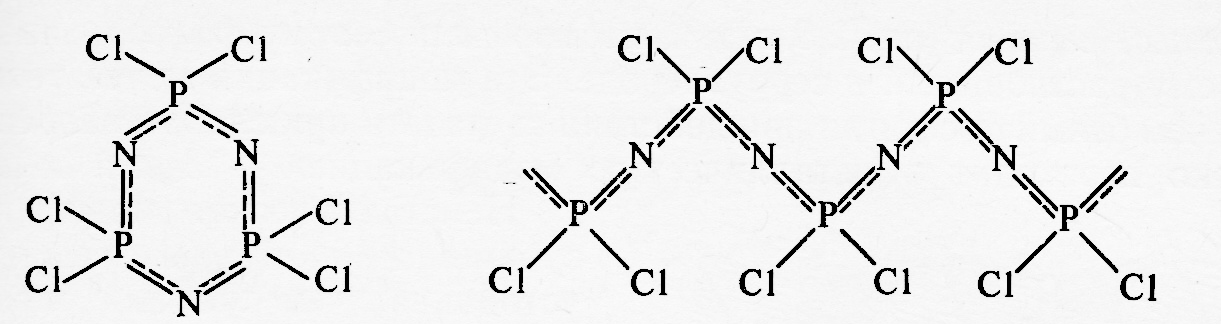
б) в)
Рис. 35. Гетероатомные полимеры, на основе цепей из атомов азота и фосфора: а)фосфонитриламин, б) циклическая форма фосфонитрилхлорида, в) полимерная форма фосфонитрилхлорида
В зависимости от природы боковых радикалов они могут обладать свойствами пластиков или каучуков. Поэтому их используют для изготовления эластомеров, которые могут эксплуатироваться при температурах вплоть до (- 200оС), а также в различных агрессивных средах. Указанные материалы могут иметь относительное удлинение 100 – 200%, а их прочность на растяжение в 12 – 30 раз больше, чем у органических эластомеров. Они устойчивы к действию компонентов топлива, маслам, не горючи и имеют высокую износостойкость.
Наибольшее число гетероатомных полимеров образуется как результат сочетания атомов различных элементов с атомами кислорода, серы и азота. В частности, как отмечалось ранее, многие гидроксиды, центральные атомы которых имеют среднее значение ПД, являются полимерными оксо-гидроксо-аквокомплексами, а на их основе возможно формирование многочисленных полимерных оксо-гидроксо-ацидо-аквокомплексов. К полимерным относятся и сульфиды элементов главных подгрупп III – V подгрупп, а также галогенды ряда элементов (рис. 36).
Рис.36. Строение полимерных сульфидов и галогенидов.
К особым видам полимеров можно отнести бороводороды (рис.37). К полимерам также относятся и многие халькогалогентды р- элементов, например состава АВС ( рис.38).
Рис. 37. Строение полимерных молекул бороводородов.
38. Строение кристалла SbSI
Большое число полимеров, обладающих разнообразными свойствами может быть получено при разложении аминокомплексов, например состава [Me(NH3)6]3+. Если от таких комплексов последовательно отщеплять молекулы аммиака, то это приводит к так называемому ацидированию, 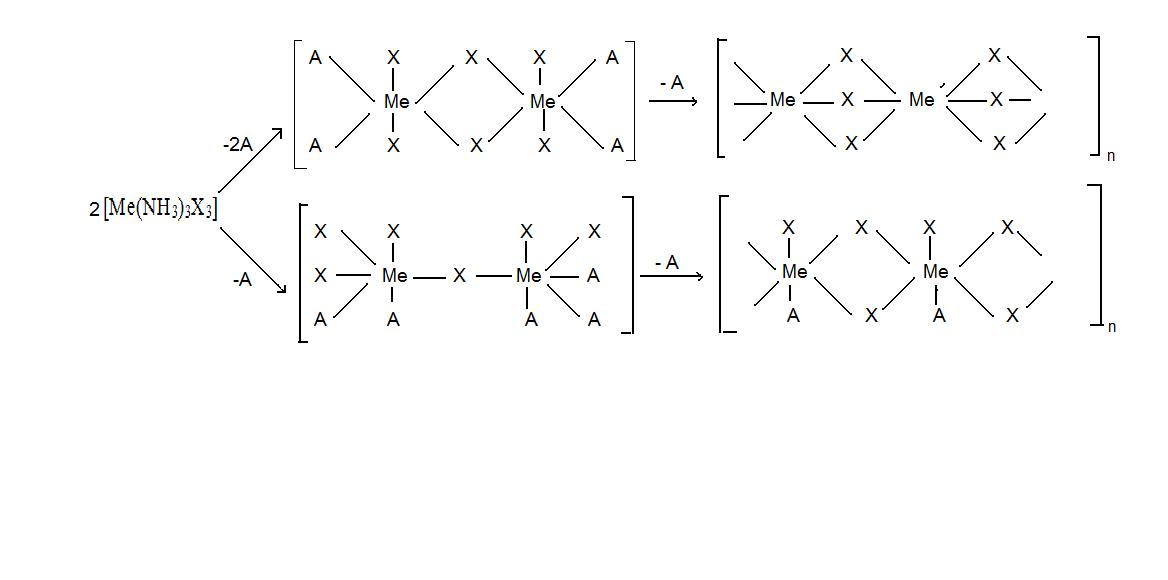 в ходе которого внешние ионы Х- занимают места молекул NH3 во внутренней сфере:
в ходе которого внешние ионы Х- занимают места молекул NH3 во внутренней сфере:
[Me(NH3)6]Х3 [Me(NH3)3Х3] + 3NH3
Если продолжить процесс отщепления NH3, то для сохранения к.ч. = 6, должна протекать полимеризация исходных мономеров (на рис. А = NH3).
Химическим способом удаления NH3 из системы может быть действие раствора щёлочи на эти комплексы.
Разнообразие видов гетероатомных комплексов иллюстрирует таблица 18.
Таблица 18. Комбинации р-элементов, образующих гетероатомные неорганические полимеры.
|
Э1 |
Э2 |
Э1 |
Э2 |
||
|
О |
N |
О |
N |
||
|
B |
+ |
+ |
P |
+ |
+ |
|
Al |
+ |
+ |
As |
+ |
+ |
|
Si |
+ |
+ |
Sb |
+ |
+ |
|
Ge |
+ |
- |
S |
+ |
+ |
|
Sn |
+ |
- |
Se |
+ |
+ |
|
Pb |
+ |
+ |
Te |
+ |
+ |
Литература
Угай Я.А.: Общая и неорганическая химия. - М.: Высшая школа, 2004
Угай Я.А.: Общая и неорганическая химия. - М.: Высшая школа, 2004
Ю.М. Коренев, А.Н. Григорьев, Н.Н. Хелиговская, К.М. Дунаева; Под ред. Ю.Д. Третьякова; Рец.: Московская гос. акад. тонкой химической технологии им. М.В. Ломоносова, В.А. Собянин, С.В. Коренев: Задачи и вопросы по общей и неорганической химии с ответами и решениями. - М.: Мир, 2014
Ахметов Н.С.: Общая и неорганическая химия. - М.: Высшая школа, 2013
Ахметов Н.С.: Общая и неорганическая химия. - М.: Высшая школа, 2013
Б.Д. Степин, Л.Ю. Аликберова, Н.С. Рукк, Е.В. Савинкина; Под ред. Б.Д. Степина: Демонстрационные опыты по общей и неорганической химии. - М.: ВЛАДОС, 2013
Бабков А.В.: Общая, неограническая и органическая химия. - М.: Дрофа, 2003
Белгородская государственная сельскохозяйственная академия; Сост.: И.И. Василенко, Н.Г. Габрук, Н.М. Шевель, Л.А. Манохина; Рец. Г.М. Фофанов: Неорганическая химия. - Белгород: БелГСХА, 2013
Гузей Л.С.: Химия. 11 класс. - М.: Дрофа, 2003
Гузей Л.С.: Химия. 8 класс. - М.: Дрофа, 2003
Гузей Л.С.: Химия. 9 класс. - М.: Дрофа, 2103
МО РФ; БелГУ, каф. неорганической химии; Сост. И.И. Олейникова: Программа промежуточной аттестации студентов по дисциплине "Химия". - Белгород: БелГУ, 2013
МО РФ; БелГУ, каф. неорганической химии; Сост. И.И. Олейникова: Учебная программа дисциплины "Химия". - Белгород: БелГУ, 2008
МО РФ; БелГУ, каф. неорганической химии; Сост. И.И. Олейникова: Учебная программа дисциплины "Химия". - Белгород: БелГУ, 2008
Плесков Ю.В.: Электрохимия алмаза. - М.: Едиториал УРСС, 2009
В.И.Фролов, Т.М. Курохтина, З.Н. Дымова и др.; Рец.: А.П. Нечаев: Практикум по общей и неорганической химии. - М.: Дрофа, 2012
Гузей Л.С.: Химия. 8 класс. - М.: Дрофа, 2012
Лидин, Р.А.: Химия для школьников старших классов и поступающих в вузы. Теоретические основы. Вопросы. Задачи. Тесты. - М.: Дрофа, 2007
Павлов Н.Н.: Общая и неорганическая химия. - М.: Дрофа, 2006
Павлов, Н.Н.: Общая и неорганическая химия. - М.: Дрофа, 2010
Рудзитис Г.Е.: Химия : Неорганическая химия. - М.: Просвещение, 2012
УМО по медицинскому и фармацевтическому образованию вузов России; ГОУ Всероссийский учебно-научно-методический Центр по непрерывному медицинскому и фармацевтическому образованию; Н.В. Головина, Н.В. Машнина, В.А. Попков, С.А. Пузаков; Под ред.: В.А.: Химия. - М.: ГОУ ВУНМЦ МЗ РФ, 2002
Гузей Л.С.: Химия. 10 класс. - М.: Дрофа, 2011
Гузей Л.С.: Химия. 11 класс. - М.: Дрофа, 2011
Зуева М.В.: Контрольные и проверочные работы по химии. 8-9классы. - М.: Дрофа, 2010
Под ред. В.Ю. Баранова; Б.М. Андреев, В.Ю. Баранов, И.А. Белов и др.: Изотопы: свойства, получение, применение. - М.: ИздАТ, 2000
1
2 2 ZrOZr.
Генеалогия неорганических фаз