ХИМИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ Е.В. СЕРЕБРЯКОВА
МИНИСТЕРСТВО ВЫСШЕГО ОБРАЗОВАНИЯ РФ
ВЯТСКИЙ
ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
Е.В. СЕРЕБРЯКОВА
ХИМИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ
Учебное пособие
Киров, 2014
Лекция 1
ВВЕДЕНИЕ
Накоплением знаний, анализом явлений и фактов занимается наука. Если в период своего зарождения наука была единой, неделимой и эта прекрасная, органически свойственная ей черта особенно ярко проявилась в энциклопедических трудах великих мыслителей древности, то позднее наступила пора дифференциации науки.
Из унитарной, стройной системы естествознания как единого целого возникли математика, физика, химия, биология и медицина, а в науках об обществе оформились история, философия, право...
Это неизбежное дробление науки, отражающее объективные процессы в развитии мира, продолжается и сегодня — появились кибернетика, ядерная физика, химия полимеров, океанология, экология, онкология и десятки других наук.
Веянием времени стала и узкая специализация ученых, целых коллективов. Конечно, это отнюдь не исключает становления и воспитания широко образованных ученых с блестящей эрудицией, и мировая наука знает немало тому примеров.
И все же вопрос закономерен — не утрачивается ли в таком случае возможность осмысления целостной картины окружающего мира, не мельчает ли порой постановка проблем, не ограничиваются ли искусственно поиски путей их решения? Особенно для тех, кто только начинает свой путь к знаниям...
Отражением этого противоречия и прямым следствием действия законов диалектики явилось встречное движение наук по пути к взаимному обогащению, взаимодействию и интеграции.
Появились математическая лингвистика, химическая физика, биологическая химия...
Что будет конкретным и конечным итогом этого непрерывного искания, постоянной смены целей и объектов исследования, предсказать пока трудно, но одно является очевидным — в конечном итоге человек достигнет прогресса и в тех областях знания, которые совсем недавно казались окутанными покровом глубокой тайны...
Одним из ярких примеров является та область науки, которая лежит на границе биологии и химии.
Что же объединяет эти научные дисциплины, в чем смысл их взаимодействия?
Ведь биология была и, пожалуй, еще долгое время будет одной из самых загадочных областей знания, и в ней остается немало белых пятен.
Химия же, напротив, относится к разряду наук наиболее устоявшихся, точных, в ней основные закономерности выяснены и проверены временем.
И тем не менее факт остается фактом — уже давно химия и биология идут навстречу друг другу.
Когда это началось, вряд ли можно сейчас установить... Попытки объяснения явлений жизнедеятельности с позиций точных наук мы находим еще у мыслителей древнегреческой и древнеримской цивилизации, более отчетливо подобные идеи формулировались в трудах выдающихся представителей научной мысли средневековья и эпохи Возрождения.
К концу XVIII в было достоверно установлено, что в основе проявления жизни лежа химические превращения веществ, порой простых, а зачастую удивительно сложных. И именно с этого периода начинается подлинная летопись о союзе двух наук, летопись, богатая ярчайшими фактами и эпохальными открытиями, фейерверк которых не прекращается и в наши дни...
Крупнейшим событием можно считать рождение органической химии, которая первоначально рассматривалась как химия веществ, встречающихся в живой природе.
На первых этапах в ней господствовали виталистические воззрения, утверждавшие, что химические соединения, выделяемые из живых организмов, не могут быть получены искусственным путем, без участия магической жизненной силы.
Сокрушительный удар сторонникам витализма был нанесен работами Ф. Вёлера, получившего типичное вещество животного происхождения — мочевину из цианата аммония. Последующими исследованиями позиции витализма были окончательно подорваны.
В середине XIX в. органическая химия определяется уже как химия соединений углерода вообще — будь то вещества природного происхождения или синтетические полимеры, красители или лекарственные препараты.
Один за другим преодолевала органическая химия барьеры, стоящие на пути к познанию живой материи.
В 1842 г. Н. Н. Зинин осуществил синтез анилина, в 1854 г. М. Бертло получил синтезом ряд сложных органических веществ, в том числе жиры.
В 1861 г. А. М. Бутлеровым впервые было синтезировано сахаристое вещество — метиленитан, к концу столетия успешно осуществляются синтезы ряда аминокислот и жиров, а начало нашего века ознаменовалось первыми синтезами белковоподобных полипептидов.
Это направление, развивавшееся стремительно и плодотворно, оформилось к началу XX в. в самостоятельную химию природных соединений.
К числу ее блистательных побед можно отнести расшифровку строения и синтез биологически важных алкалоидов, терпеноидов, витаминов и стероидов, а вершинами ее достижений в середине нашего века надо считать полные химические синтезы хинина, стрихнина, резерпина, пенициллина и простагландинов.
Биологическими проблемами занимаются сегодня десятки наук, в которых тесно переплетаются идеи и методы биологии, химии, физики, математики и других областей знания.
Арсенал используемых биологией средств огромен. Именно в этом — один из источников ее бурного прогресса, основа достоверности ее выводов и суждений.
Пути биологии и химии в познании механизмов жизнедеятельности пролегают рядом, и это естественно, ибо живая клетка — настоящее царство больших и малых молекул, непрерывно взаимодействующих, возникающих и исчезающих...
Здесь находит сферу приложения и одна из новых наук — биоорганическая химия.

Биоорганическая химия — наука, которая изучает связь между строением органических веществ и их биологическими функциями.
Объектами изучения являются, такие как: биополимеры, витамины, гормоны, антибиотики, феромоны, сигнальные вещества, биологически активные вещества растительного происхождения, а также синтетические регуляторы биологических процессов (лекарственные препараты, пестициды и др.), биорегуляторы и отдельные метаболиты.
Являясь разделом (частью) органической химии эта наука также изучает соединения углерода.
В настоящее время насчитывается – 16 млн органических веществ.
Причины многообразия органических веществ:
1) Соединения атомов углерода (С) могут взаимодействовать друг с другом и другими элементами периодической системы Д. И. Менделеева. При этом образуются цепи и циклы.
2) Атом углерода может находиться в трех разных гибридных состояниях. Тетраэдрическая конфигурация атома С плоскостная конфигурация атома С.
3) Гомология – это существование веществ с близкими свойствами, где каждый член гомологического ряда отличается от предыдущего на группу – СН2—.
4) Изомерия – это существование веществ, имеющих одинаковый качественный и количественный состав, но различное строение.
А) M. Бутлеров (1861 г.) создал теорию строения органических соединений, которая и по сей день служит научной основой органической химии.
Б) Основные положения теории строения органических соединений:
1) атомы в молекулах соединены друг с другом химическими связями в соответствии с их валентностью;
2) атомы в молекулах органических соединений соединяются между собой в определенной последовательности, что обусловливает химическое строение молекулы;
3) свойства органических соединений зависят не только от числа и природы входящих в их состав атомов, но и от химического строения молекул;
4) в молекулах существует взаимное влияние как связанных, так и непосредственно друг с другом не связанных атомов;
5) химическое строение вещества можно определить в результате изучения его химических превращений и, наоборот, по строению вещества можно охарактеризовать его свойства.
Итак, объектами изучения биоорганической химии являются:
1) биологически важные природные и синтетические соединения: белки и пептиды, нуклеиновые кислоты, углеводы, липиды,
2) биополимеры смешанного типа — гликопротеины, нуклеопротеины, липопротеины, гликолипиды и т. п.; алкалоиды, терпеноиды, витамины, антибиотики, гормоны, простагландины, ростовые вещества, феромоны, токсины,
3) а также синтетические лекарственные препараты, пестициды и др.
Биополимеры – высокомолекулярные природные соединения, которые являются основой всех организмов. Это белки, пептиды, полисахариды, нуклеиновые кислоты (НК), липиды.
Биорегуляторы – соединения, которые химически регулируют обмен веществ. Это витамины, гормоны, антибиотики, алкалоиды, лекарственные препараты и др.
Знание строения и свойств биополимеров и биорегуляторов позволяет познать сущность биологических процессов. Так, установление строения белков и НК позволило развить представления о матричном биосинтезе белка и роли НК в сохранении и передаче генетической информации.
Основная задача биоорганической химии – выяснение взаимосвязи структуры и механизма действия соединений.

Итак, из сказанного понятно, что биоорганическая химия – это научное направление, сложившееся на стыке ряда отраслей химии и биологии.
В настоящее время она превратилась в фундаментальную науку. По существу она является химическим фундаментом современной биологии.
Разрабатывая основополагающие проблемы химии живого мира, биоорганическая химия способствует решению задач получения практически важных препаратов для медицины, сельского хозяйства, ряда отраслей промышленности.
Основные задачи:
- выделение в индивидуальном состоянии изучаемых соединений с помощью кристаллизации, перегонки, различных видов хроматографии, электрофореза, ультрафильтрации, ультрацентрифугирования, противоточного распределения и т. п.;
- установление структуры, включая пространственное строение, на основе подходов органической и физико-органической химии с применением масс-спектрометрии, различных видов оптической спектроскопии (ИК, УФ, лазерной и др.), рентгеноструктурного анализа, ядерного магнитного резонанса, электронного парамагнитного резонанса, дисперсии оптического вращения и кругового дихроизма, методов быстрой кинетики и т. п. в сочетании с расчетами на ЭВМ;
- химический синтез и химическая модификация изучаемых соединений, включая полный синтез, синтез аналогов и производных,— с целью подтверждения структуры, выяснения связи строения и биологической функции, получения практически ценных препаратов;
- биологическое тестирование полученных соединений in vitro и in vivo.
Решение основных проблем Б. х. важно для дальнейшего прогресса биологии. Без выяснения строения и свойств важнейших биополимеров и биорегуляторов нельзя познать сущность жизненных процессов, а тем более найти пути управления такими сложными явлениями, как:
-размножение и передача наследственных признаков,
- нормальный и злокачественный рост клеток,-
-иммунитет, память, передача нервного импульса и многое др.
В то же время изучение высокоспециализированных биологически активных веществ и процессов, протекающих с их участием, может открыть принципиально новые возможности для развития химии, химической технологии и техники.
К проблемам, решение которых связано с исследованиями в области Б. х., относятся:
- создание строго специфичных высокоактивных катализаторов (на основе изучения строения и механизма действия ферментов),
- прямое превращение химической энергии в механическую (на основе изучения мышечного сокращения),
-использование в технике химических принципов хранения и передачи информации, осуществляемых в биологических системах, принципов саморегулирования многокомпонентных систем клетки в первую очередь избирательной проницаемости биологических мембран, и многое др.
Перечисленные проблемы лежат далеко за пределами собственно Б. х.; однако она создает основные предпосылки для разработки этих проблем, обеспечивая главные опорные пункты для развития биохимических исследований, относящихся уже к области молекулярной биологии. Широта и важность решаемых проблем, разнообразие методов и тесная связь с другими научными дисциплинами обеспечили быстрое развитие Б. х.
Биоорганическая химия сформировалась в самостоятельную область в 50-х гг. 20 в.
В этот же период это направление начало делать первые шаги в Советском Союзе.

Заслуга в этом принадлежала академику Михаилу Михайловичу Шемякину.
Тогда ему оказали решительную поддержку руководители Академии наук А. Н. Несмеянов и Н. Н. Семенов, и уже в 1959 г. в системе АН СССР был создан базовый институт химии природных соединений АН СССР , который он возглавил с момента его создания (1959) до 1970 года. С 1970 по 1988 год , после смерти Михаила Михайловича Шемякина, институт возглавил его ученик и последователь академик Ю. А. Овчинников. «Развиваясь в недрах органической химии с самого начала ее зарождения как науки, она не только питалась и питается всеми представлениями органической химии, но и сама непрерывно обогащает последнюю новыми идеями, новым фактическим материалом принципиальной важности, новыми методами» – говорил академик, крупный ученый в области органической химии Михаил Михайлович Шемякин (1908-1970)»

В 1963 г. организовано Отделение биохимии, биофизики и химии физиологически активных соединений АН СССР. Соратниками М. М. Шемякина в этой деятельности, а порой и борьбе, были академики А. Н. Белозерский и В. А. Энгельгардт; уже в 1965 г. Академик А. Н. Белозерский основал Межфакультетскую лабораторию биоорганической химии МГУ, которая сейчас носит его имя.
Методы и с с л е д о в а н и я : основной арсенал составляют методы органической химии, однако для решения структурно-функциональных задач привлекаются и разнообразные физические, физико-химические, математические и биологические методы.

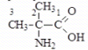
Аминокислоты (аминокарбоновые кислоты) - являются бифункциональными соединениями, которые содержат в молекуле две реакционноспособные группы: карбонильные (–СООН), аминогруппу (–NH2), -атом углерода (в центре) и радикал (различается у всех -аминокислот).
Аминокислоты могут рассматриваться как производные карбоновых кислот, в которых один или несколько атомов водорода заменены на аминные группы.
Аминокислоты (кроме глицина) существуют в двух стереоизомерных формах – L и D, вращающих плоскость поляризации света соответственно влево и вправо.
Все живые организмы синтезируют и усваивают только L-аминокислоты, а D-аминокислоты для них либо безразличны, либо вредны. В естественных белках встречаются преимущественно -аминокислоты, в молекуле которых аминогруппа присоединена к первому атому (-атому) углерода; у -аминокислот аминогруппа находится при втором атоме углерода.
Аминокислоты являются мономерами, из которых строятся полимерные молекулы – протеины, или белки.
Как уже отмечалось ранее, практически все природные -аминокислоты оптически активны (за исключением глицина) и относятся к L-ряду. Это означает, что в проекции Фишера, если внизу расположить заместитель, а вверху карбоксильную группу, то аминогруппа будет находиться слева.
Это, разумеется, не означает, что все природные аминокислоты вращают плоскость поляризованного света в одну и ту же сторону, поскольку направление вращения определяется свойствами всей молекулы, а не конфигурацией его асимметрического атома углерода. Большая часть природных аминокислот имеет S-конфигурацию (в том случае, когда в ее состав входит один асимметрический атом углерода).
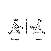
Некоторые микроорганизмы синтезируют аминокислоты D-ряда. Такие аминокислоты называют “неприродными”.
Конфигурацию протеиногенных аминокислот соотносят с D - глюкозой; такой подход предложен Э. Фишером в 1891 г. В пространственных формулах Фишера заместители у хирального С-2 атома занимают положение, которое соответствует их абсолютной конфигурации (это было доказано через 60 лет).
На рисунке приведены пространственные формулы D- и L-аланина.
Все аминокислоты, за исключением глицина, оптически активны благодаря хиральному строению.
Энантиомерные формы, или-оптические антиподы, имеют различные показатели преломления (круговое двулучепреломление) и различные коэффициенты молярной экстинкции (круговой дихроизм) для лево и право циркулярно поляризованных компонент линейно-поляризованного света. Они поворачивают плоскость колебаний линейного поляризованного света на равные углы, но в противоположных направлениях. Вращение происходит так, что обе световые составляющие проходят оптически активную среду с различной скоростью и при этом сдвигаются по фазе.
По углу вращения а, определенному на поляриметре, можно определить удельное вращение [a] D.
ИЗОМЕРИЯ АМИНОКИСЛОТ

1)Изомерия углеродного скелета
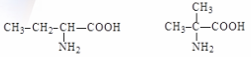
2) Изомерия положения аминогруппы
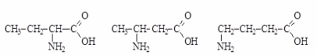
3) Межклассовая изомерия
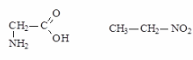
В двумерном изображении для D- И L-изомеров принят определенный порядок расположения заместителей. У D-аминокислоты наверху изображают карбоксильную группу, далее следуют по часовой стрелке аминогруппа, боковая цепь и атом водорода (см. ниже). У L-аминокислоты принят обратный порядок расположения заместителей, причем боковая цепь всегда стоит внизу.

По рациональной номенклатуре названия аминокислот строятся следующим образом: за основу выбирают травиальное название соответствующей карбоновой кислоты, к которому добавляют приставку «амино-». Положение аминогруппы обозначают греческими буквами, для чего углеродную цепь аминокислоты нумеруют, начиная с атома углерода, соседнего с карбоксильной группой.
Например:
|
|
|
|
|
– аминопропионовая кислота |
- аминомасляная кислота |
– аминомасляная кислота |
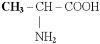
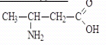
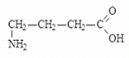
По номенклатуре ЮПАК названия аминокислот строятся следующим образом: за основу выбирают самую длинную цепь, содержащую карбоксильную и аминогруппы, NH2 – группа обозначается приставкой «амино-», ее положение обозначается цифрой, причем нумерация начинается с атома углерода карбоксильной группы. К названию основы добавляется окончание «-овая» и слово кислота.
|
Например:
3-аминобутановая кислота |
2-амино-2-метилпропановая кислота |
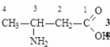
Название – аминокислот могут быть построены по заместительной номенклатуре, но чаще используются их тривиальные названия
|
|
|
|
|
2-аминопропановая к-та; –аминопропионовая к-та; - аланин |
2-аминобутандионовая (аминоянтарная) к-та, аспарагиновая к-та |
2-амино-3-меркаптопропановая кислота, цистеин |
|
|
Формула аминокислоты |
Тривиальное название |
|
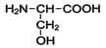
2-амино-З-гидроксипропановая кислота |
|
Серин |
|
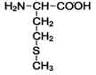
2-амино- 4-метилтиомасляная кислота |
|
Метионин |

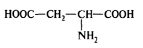
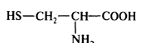
Все природные аминокислоты имеют тривиальные названия:
|
|
|
|
|
глицин |
аланин |
аспарагиновая к-та |
|
|
|
|
|
лизин |
фенилаланин |
|
|
|
|
|
|
гистидин |
серин |
|
|
|
|
|
|
цистеин |
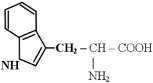
триптофан |
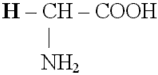
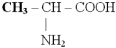
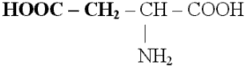
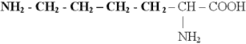
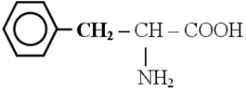
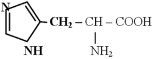
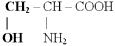
КЛАССИФИКАЦИЯ АМИНОКИСЛОТ
По числу карбоксильных и аминогрупп:
1) Моноаминомонокарбоновые кислота – она же Глицин (-аминопропионовая кислота-аланин, – аминоизовалериановая кислота – валин, – аминоизокапроновая кислота – лейцин):
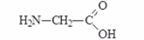
Глицин
(аминоуксусная кислота)
2) Моноаминодикарбоновые кислоты - L(+) – аспарагиновая или -аминоянтарная кислота, (глутаминовая кислота, аспарагин).
Двухосновные аминокислоты обладают не нейтральной, как одноосновные, а кислой реакцией. В остальном реакции двухосновных аминокислот аналогичны одноосновных - аминокислот.
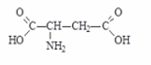 Аспарагиновая кислота, (- аминоянтарная кислота)
Аспарагиновая кислота, (- аминоянтарная кислота)
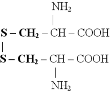
3) Диаминомонокарбоновые кислоты - , – диаминокапроновая кислота (L-(+)-лизин), ( еще- диаминовалериановая кислота – L-орнитин)
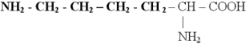 Лизин, ( –диаминокапроновая кислота)
Лизин, ( –диаминокапроновая кислота)

По характеру углеводородного радикалы:
1) алифатические (радикал является остатком углеводорода);
2) ароматические (в состав радикала входит остаток ароматического углеводорода);
3) гетероциклические (в состав радикала входит гетероцикл).
По характеру углеводородного радикала

1) - оксиаминокислоты (содержат в радикале гидроксильную группу)
2) серосодержащие аминокислоты (в состав радикала входит один или несколько атомов серы)
V По положению аминогруппы

1)- – аминокислоты;
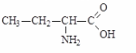
–аминомасляная кислота; этилглицин;
2-аминобутановая.
2) - аминокислоты
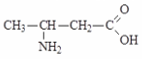
– аминомасляная кислота, 3-аминобутановая
3) - аминокислоты
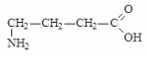
– аминомасляная кислота
4-аминобутановая
Гамма-аминомасляная кислота (ГАМК) - принимает участие в обменных процессах происходящих в головном мозге, является нейромедиатором. В медицинской практике под названием гаммалон, иди аминалон, применяется при лечении нервнопсихических заболеваний. Структура ГАМК лежит в основе транквилизатора фенибута. Важное значение для медицины имеют производные циклической формы ГАМК — ее лактама (пирролидон-2). В частности, полимер — поливинилпирролидон — эффективный заменитель плазмы крови.
Бета-гидроксимасляная кислота CH3-CН(ОН)-CН2-CООН как промежуточный продукт окисления жирных кислот накапливается в организме у больных сахарным диабетом, являясь, в свою очередь, предшественником ацетоуксусной кислоты.
-аминомасляная кислота - принимает участие в обменных процессах происходящих в головном мозге, является нейромедиатором. В медицинской практике под названием гаммалон, иди аминалон, применяется при лечении нервнопсихических заболеваний. Структура ГАМК лежит в основе транквилизатора фенибута.
V По путям биосинтеза

Пути биосинтеза протеиногенных аминокислот разноплановы.
Одна и та же аминокислота может образовываться разными путями. К тому же совершенно различные пути могут иметь очень похожие этапы.
Тем не менее, имеют место и оправданы попытки классифицировать аминокислоты по путям их биосинтеза.
Существует представление о следующих биосинтетических семействах аминокислот: аспартата, глутамата, серина, пирувата и пентоз.
Не всегда конкретную аминокислоту можно однозначно отнести к определённому семейству; делаются поправки для конкретных организмов и учитывая преобладающий путь.
По семействам аминокислоты обычно распределяют следующим образом:
- семейство аспартата: аспартат, аспарагин, треонин, изолейцин, метионин, лизин;
- семейство глутамата: глутамат, глутамин, аргинин, пролин;
-семейство пирувата: аланин, валин, лейцин;
- семейство серина: серин, цистеин, глицин;
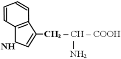
- семейство пентоз: гистидин, фенилаланин, тирозин, триптофан;
- иногда выделяют в семейство шикимата: фенилаланин, тирозин, триптофан.
V По способности организма синтезировать из предшественников

Существует 22 аминокислоты, среди которых выделяют незаменимые, условно незаменимые и незаменимые:
1) незаменимыми - валин, изолейцин, лейцин, треонин, метионин, лизин, (пирролизин-22 аминокислота) фенилаланин, триптофан, аргинин, гистидин;
2) - заменимые - глицин, аланин, пролин, серин, цистеин, аспарагиновая кислота, аспарагин, глутамин, глутаминовая кислота, тирозин.
3) - условно незаменимые- тирозин, цистеин (селеноцистеин - 21 аминокислота)
Аминокислоты, синтезируемые в организме, называются заменимыми, а те которые не могут синтезироваться, незаменимыми.
В зависимости от вида животных выделяют от 8-10 незаменимых аминокислот: валин, лейцин, аргинин триптофан, гистидин, фенилаланин.
Для птицы выделяют еще и глицин.
Соотношение и содержание заменимых и незаменимых аминокислот определяют полноценность белков. Полноценными являются белки животного происхождения. Растительные белки обычно содержат мало незаменимых аминокислот и их относят к неполноценным.
У растений все необходимые аминокислоты синтезируются из первичных продуктов фотосинтеза.
Однако классификация аминокислот на заменимые и незаменимые не лишена недостатков.
К примеру, тирозин является заменимой аминокислотой только при условии достаточного поступления фенилаланина.
Для больных фенилкетонурией тирозин становится незаменимой аминокислотой.
Аргинин синтезируется в организме человека и считается заменимой аминокислотой, но в связи с некоторыми особенностями его метаболизма при определённых физиологических состояниях организма может быть приравнен к незаменимым.
Гистидин также синтезируется в организме человека, но не всегда в достаточных количествах, потому должен поступать с пищей.
Лекция 2
СПОСОБЫ ПОЛУЧЕНИЯ АМИНИКИСЛОТ
Химический синтез аминокислот

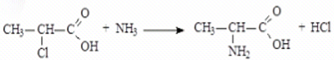
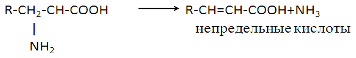
1) Из галогенопроизводных карбоновых кислот - действие избытка аммиака на – галогенокислоты.
При взаимодействии -хлор- или -бромкарбоновых кислот с избытком водного, спиртового или жидкого аммиака, при температуре 40-50 С, в результате нуклеофильного замещения образуются соответствующие -аминокислоты

Монохлорпропионовая к-та Аланин
(-аминопропионовая кислота)
CH2–COOH CH2– COOH
| |
Cl NH2
Монохлуксусная Глицин
к-та (-аминоуксусная к-та)
2) взаимодействием галогенокарбоновых кислот с аммиаком:

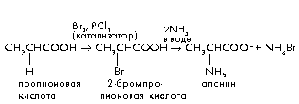
В лабораторных условиях химический синтез аминокислот осуществляют при взаимодействии - галогенокарбоновых кислот с аммиаком.
Исходные -галогенокислоты обычно получают по реакции Гелля–Фольгарда–Зелинского:
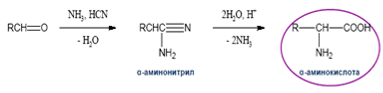
По этому методу из аммиака, альдегидов и синильной кислоты получают -аминонитрилы, гидролиз которых дает -аминокислоты.
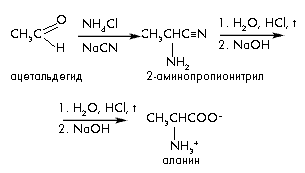
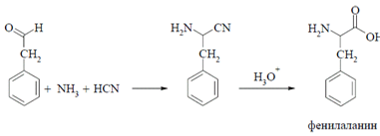
3) Синтез Штреккера-Зелинского (из аммиака, альдегидов и синильной кислоты получают -аминонитрилы).
В синтезе Штреккера-Зелинского альдегид превращают в -аминокислоту с удлинением углеродной цепи на один атом углерода.
По этому методу из альдегидов или кетонов действием аммиака и синильной кислоты с последующим гидролизом получают -аминонитрилы, гидролиз которых дает -аминокислоты.
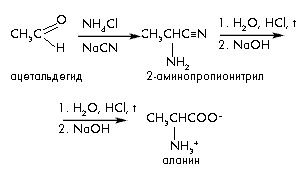
Процесс проходит в две стадии:
- на первой стадии в результате реакции альдегида с NH4Cl и NaCN получают -аминонитрил;
- на второй – при гидролизе нитрильной группы -аминонитрила получают аминокислоту:
Получение фенилаланина:
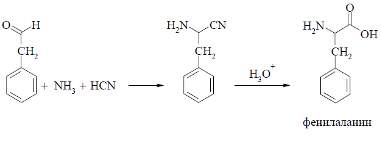
Если в недавнем прошлом -аминокислоты получали в небольших количествах, преимущественно для научных исследований, то в настоящее время налажено их многотоннажное промышленное производство. Это связано, например, с тем, что -аминокислоты являются необходимым компонентом комбикормов и синтетической пищи на углеводной основе.
4) Очень распространен способ получения -аминокислот с использованием малонового эфира:

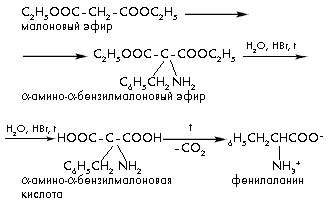
При этом один из атомов водорода метиленовой группы малонового эфира замещается на аминогруппу, а другой – на соответствующий требуемой аминокислоте углеводородный радикал. В результате гидролиза полученного диэтилового эфира и последующего декарбоксилирования дикислоты получают нужную аминокислоту.
5) Еще один способ синтеза аминокислот заключается в восстановительном аминировании (восстановлении водородом в присутствии аммиака) -оксокарбоновых кислот:

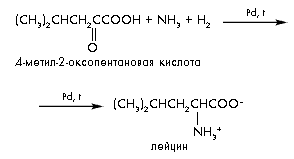
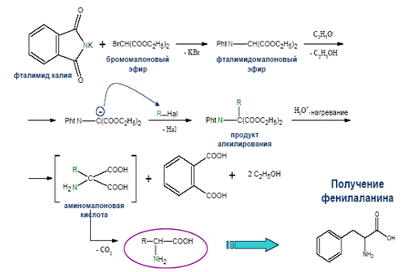
6) Аминирование галогенокислот фталимидом

Аминокислоты можно получать химическим синтезом, с последующей трансформацией с помощью микроорганизмов или ферментов, выделенных из них гидролизом природных белков, микробиологическим синтезом.
Более 60% всех производимых промышленностью чистых препаратов аминокислот получают путем микробиологического синтеза. На втором месте по объему производства находится химический синтез.
Синтез целого ряда аминокислот химическим путем хорошо изучен и введен в производство. Во многих случаях такое производство экономически выгодно. Но в процессе химического синтеза преимущественно образуются рацематы – смесь D – и L – форма аминокислот.
D-форма не имеет физиологической ценности для человека и животных: она не включается в обмен веществ и не усваивается. Очистка продукта от D-формы приводит к значительным экономическим издержкам и усложнению производства.
Преимущественно химическим путем в промышленности производится глицин, DL-метионин, L-фенилаланин, L-валин, L-треонин, L-триптофан.
Основным недостатком химического синтеза является получение смеси аминокислот, состоящей из изомеров, относящихся как к D – так и к L – ряду, тогда как биологической активностью в организме человека и животных обладают лишь L-формы. D – формы аминокислот не превращаются ферментными системами этих организмов, а некоторые из них токсичны для человека и животных. Исключением является аминокислота – метионин, у которой биологически активные как D – так и к L – формы, в связи с чем данная аминокислота производится преимущественно методом химического синтеза.
Сначала происходит окислительное дезаминирование с помощью специфической D-аминокислотной оксидазы. Затем полученная - кетокислота стереоспецифически переаминируется в L-аминокислоту. Вообще говоря, HAK можно заменить промежуточными продуктами их биосинтеза, например соответствующими кетокислотами.
Технологически получение аминокислот гидролизом белков экономически менее выгодно, поэтому не получило широкого распространения.
Физиологически активные L-формы получают в промышленном масштабе путем кислотного и щелочного гидролиза природных белков. Наиболее подходящим сырьем для данного процесса являются отходы различных производств, в том числе непищевых (например, кератинсодержащие отходы). Но этот метод имеет определенные недостатки: высокая стоимость процесса гидролиза, сложность удаления необходимой аминокислоты из смеси аминокислот гидролизата, разрушение части аминокислот в процессе гидролиза и ограниченность сырьевых ресурсов. Преимуществом способа является трансформация отходов непищевых производств в полезный продукт.
Производство аминокислот из белкового гидролизата, как способ получения L-аминокислот в настоящее время имеет лишь ограниченное значение, хотя по-прежнему является основным для производства L-серина, L-пролина, L-оксипролина и L-тирозина. Он не подходит для крупномасштабного производства аминокислот.
Промышленное использование ферментов для производства L-аминокислот началось почти 40 лет назад в Японии с разрешением использования N-ацетил-DL-аминокислот, образованных с помощью иммобилизованной ацилазы. Это послужило началом перспективного метода получения аминокислот, которое известно, как разделение рацематов. Метод осуществляется путем ассиметричного гидролиза производных аминокислот с использованием микроорганизмов, обладающих специфической L-ацилазной, L- амидазной, L- эстеразной активностью.
Ферментативное разделение рацематов аминокислот с L-ацилазами основано на избирательном гидролизе ацилированных производных L-аминокислот. При отщеплении ацильной группы L-аминокислоты становятся более растворимыми и легко отделяются от малорастворимых ацилированных D-аминокислот. Не прореагировавшие производные D-аминокислот могут быть подвергнуты рацемизации и вновь использованы для ферментативного разделения.
Для производства L-метионина используется метод, при котором применяется ацилаза, выделенная из микроорганизмов Aspergillus оryzae, а процес осуществляется в ферментном мембранном реакторе (ФМР). Ежегодно проводится получение нескольких сотен тонн L-метионина и L-валина с использованием ФМР технологии.
Ферментативный синтез аминокислот основывается на процессах с использованием выделенных в индивидуальном виде ферментов, как правило, закрепленных (иммобилизованных) на инертном носителе.Процесс получения аминокислот заключается в синтезе предшественника аминокислоты и последующей его трансформации в целевую аминокислоту с использованием либо выделенных ферментов, либо микроорганизмов.
Преимущественно ферментативным путем производится L-аспарагиновая кислота. Аспартаза в присутствии аммиака катализирует прямое преобразование фумаровой кислоты в L-аспартат, который нужен в больших количествах для подсластителя аспартама. L-аспартат является также исходным материалом для ферментативного производства L-аланина с использованием иммобилизованной аспартат--декарбоксилазы.
Для L-цистеина, который ранее производился главным образом путем электрохимического восстановления L-цистина полученного гидролизом белков, существует промышленный ферментативный процесс, в котором производная тиазолина DL-2-амино-2-тиазолин-4-карбоновая кислота (АТК) превращается в L-цистеин с помощью трех ферментов (L-ATC гидролазы, S-карбамоил-L-цистеин гидролазы и АТК рацемазы), выделенных из Pseudomonas thiazolinophilum.
Ферментативные методы получения аминокислот имеют ряд преимуществ:
• Высокая концентрация веществ в перерабатываемых смесях приводит к значительному уменьшению габаритов используемого оборудования, а также к упрощению процессов выделения и очистки полупродуктов и целевых продуктов синтеза.
• Отсутствие опасности заражения технологической линии посторонними микроорганизмами и, как следствие, возможность проведения процесса в нестерильных условиях (но требования к чистоте исходного сырья и технологических линий при работе с ферментами высокие).
Широкое применение ферментов в крупномасштабном производстве ограничено их труднодоступностью и высокой стоимостью, низкой стабильностью и чувствительностью даже в иммобилизованном виде ко многим внешним факторам.
Микробиологический метод получения аминокислот, наиболее распространенный в настоящее время, основан на способности микроорганизмов синтезировать все L-аминокислоты, а в определенных условиях – обеспечивать их сверхсинтез. Биосинтез аминокислот в микробных клетках протекает в виде так называемых свободных аминокислот или «пула аминокислот», из которого в процессах конструктивного метаболизма синтезируются клеточные макромолекулы.
Пути синтеза большинства аминокислот взаимосвязаны. При этом одни аминокислоты являются предшественниками для биосинтеза других.
Синтез каждой аминокислоты в микробных клетках реализуется в строго определенных количествах, обеспечивающих образование последующих аминокислот, и находится под строгим генетическим контролем. Контроль осуществляется по принципу обратной связи на уровне генов, ответственных за синтез соответствующих ферментов (репрессия), и на уровне самих ферментов, которые в результате избытка образующихся аминокислот могут изменять свою активность (ретроингибирование). Данный механизм контроля исключает перепроизводство аминокислот и также препятствует их выделению из клеток в окружающую среду. Чтобы добиться сверхсинтеза отдельных аминокислот, нужно обойти или изменить данный контрольный механизм их синтеза. Для первого пути возможно использование природных «диких» штаммов, в этом случае существенны условия ферментации, так как добиться дисбаланса в системе синтеза аминокислот можно путем изменения ряда основных факторов среды (концентрация основного субстрата, рН, соотношение макро- и микроэлементов в среде и др.). Изменение контрольного механизма синтеза аминокислот осуществляется генетическими методами. При этом получают мутантные организмы: ауксотрофные и регуляторные мутанты.
Ауксотрофные мутанты – это организмы, утратившие способность к синтезу одной или нескольких аминокислот. Их используют в тех случаях, когда необходимо синтезировать аминокислоты, являющиеся конечными продуктами разветвленных цепей метаболических реакций аминокислот. Например, для получения L-лизина, L-треонина, L-метионина или L-изолейцина, для которых общим предшественником является L-аспартат, применяют мутанты, ауксотрофные по гомосерину или треонину и гомосерину. Ауксотрофные мутанты не способны образовывать ингибиторы соответствующего метаболического пути, работающие по принципу отрицательной обратной связи из-за отсутствия определенной ключевой ферментативной реакции. Поэтому при выращивании такого штамма микроорганизмов в среде с минимальной концентрацией необходимого ингредиента (аминокислоты) они способны на суперпродукцию аминокислоты-предшественника.
Регуляторные мутанты – мутанты с частично нарушенной регуляцией биосинтеза. Регуляторные мутанты отбирают по устойчивости к аналогам аминокислот либо среди ревертантов ауксотрофов. Аналоги аминокислот выступают в роли искусственных ингибиторов ферментов, работающих по принципу обратной связи, одновременно обеспечивая биосинтез требуемых аминокислот и подавляя процесс их включения в белки.
В последние годы для получения новых эффективных штаммов продуцентов аминокислот стали применять новейшие методы биотехнологии. Методы генетической инженерии позволяют повышать количество генов биосинтеза путем их клонирования на плазмидах. Это приводит к увеличению количества ферментов, ответственных за синтез аминокислот, следовательно, повышает выход целевого продукта. Клонирование генов системы синтеза аминокислот в клетки микроорганизмов с иным, по сравнению с донорским организмом, типом питания позволяет расширять сырьевую базу и заменять дорогостоящие сахаросодержащие субстраты более дешевыми.
До сих пор большинство штаммов-продуцентов BCAA (ВСАА - от англ. Branched-chain amino acids: L-валин, L-лейцин и L-изолейцин) были разработаны путем случайного мутагенеза. Этот классический подход был успешным, как и для других продуцентов аминокислот, но он имеет некоторые недостатки. Генетические изменения, вызванные мутагенезом, могут касаться тех частей генетического аппарата клетки, которые непосредственно не связаны с биосинтезом аминокислоты, в результате чего могут произойти нежелательные изменения в клеточной физиологии. Очень трудно осуществить дальнейшее улучшение штаммов со случайными мутациями. Лучшим решением этой проблемы является конструирование штаммов-продуцентов аминокислот с использованием методов рациональной метаболической инженерии. Чаще всего это осуществляется путем блокирования конкурирующего пути и с помощью гиперэкспрессии генов биосинтеза.
Аминокислоты L-фенилаланин и L-цистеин, которые ранее изготавливались в основном с помощью ферментов, теперь могут быть получены более экономически эффективным путем ферментации с использованием штаммов Escherichia coli и, таким образом, стать более доступными для растущего рынка. Почти все протеиногенные аминокислоты, за немногими исключениями, могут быть изготовлены промышленным способом специально разработанными мутантными штаммами Corynebacterium glutamicum и E. coli.
ХИМИЧЕСКИЕ СВОЙСТВА АМИНОКИСЛОТ

В растворе аминокислоты могут выступать в роли как кислот, так и оснований, т. е. они являются амфотерными соединениями. Карбоксильная группа -СООН способна отдавать протон, функционируя как кислота, а аминная - NH2 — принимать протон, проявляя таким образом свойства основания.
1)Аминокислоты взаимодействуют с кислотами:
NH2 – CH2 – COOH + HCI HCI• NH2 – CH2 –COOH
(хлороводородная соль глицина)
2)Аминокислоты взаимодействуют с щелочами:
NH2 —CH2 —COOH + NaOH NH2 —CH2 —COONa + H2O
(натриевая соль глицина)
3) Дезаминирование азотистой кислотой:
H2N-CH(R)-COOH + HNO2 HO-CH(R)-COOH + N2+ H2O
Подобно первичным аминам, аминокислоты реагируют с азотистой кислотой, при этом аминогруппа превращается в гидроксогруппу, а аминокислота — в гидроксикислоту:
Измерение объема выделившегося азота позволяет определить количество аминокислоты (метод Ван-Слайка).
4) Реакция этерификации (со спиртами в присутствии газообразного хлороводорода (НСI)
H2N-CH(R)-COOH + R'OH H2N-CH(R)-COOR' + Н2О.
Аминокислоты могут реагировать со спиртами в присутствии газообразного хлороводорода, превращаясь в сложный эфир (точнее, в хлороводородную соль эфира).
Сложные эфиры аминокислот не имеют биполярной структуры и являются летучими соединениями.
Реакция этерификации – реакция между спиртом и кислотой с выделением воды и образованием сложного эфира. Эта реакция имеет внешнюю аналогию с реакцией нейтрализации кислоты щелочью, хотя сложные эфиры по свойствам нисколько не напоминают соли.
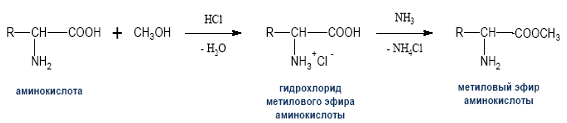
NH2–CH2– COOH+CH3OH H2O+NH2–CH2 COOCH3
(метиловый эфир глицина)
Реакция этерификации – реакция между спиртом и кислотой с выделением воды и образованием сложного эфира. Эта реакция имеет внешнюю аналогию с реакцией нейтрализации кислоты щелочью, хотя сложные эфиры по свойствам нисколько не напоминают соли.
5) Алкилирование аминогруппы:

Реакциями алкилирования называют реакции, включающие замену атома водорода органического соединения алкильным радикалом.
Алкилы (алкильные радикалы) — одновалентные радикалы насыщенных углеводородов (алканов), например метил -СН3 — это радикал метана CH4, этил -C2H5 — радикал этана C2H6.
6) Превращение аминокислот в различных средах
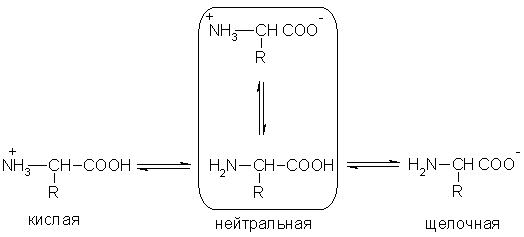
1) Аминокислоты - это органические амфотерные соединения. Они содержат в составе молекулы две функциональные группы противоположного характера: амино группу с основными свойствами и карбоксильную группу с кис лотными свойствами. Аминокислоты реагируют как с кислотами, так и с основаниями:
2) При растворении аминокислот в воде карбоксильная группа отщепляет ион водорода, который может присоединиться к ами ногруппе. При этом образуется внутренняя соль, молекула которой представляет собой биполярный ион:
+NH3– CH COO —
|
R
3) Кислотно-основные превращения аминокислот в различных средах можно изобразить следующей общей схемой:
4) Водные растворы аминокислот имеют нейтральную, щелочную или кислую среду в зависимости от количества функциональных групп.
Так, глутаминовая кислота образует кислый раствор (две группы -СООН, одна -NH2), лизин - щелочной (одна группа -СООН, две -NH2).
8) Важнейшее свойство аминокислот — их способность к конденсации с образованием пептидов.

Аминогруппа одной аминокислоты может взаимодействовать с карбоксильной группой второй, образуя пептиды (соединения, состоящие из остатков аминокислот, связанных пептидной связью):
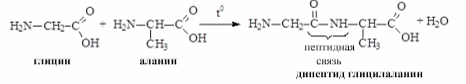
Дипептид имеет свободные –NH2 и –COOH группы и может взаимодействовать еще с одной молекулой аминогруппы, образуя трипептид, затем еще с одной молекулой – тетрапептид и т.д. При соединении друг с другом большого числа аминокислот образуются полипептиды.
Полипептидная цепь – основа белковых молекул, белки – это природные полипептиды.
ХИМИЧЕСКИЕ СВОЙСТВА АМИНОКИСЛОТ

Аминокислоты – амфотерные соединения, они могут проявлять как кислотные свойства, обусловленные наличием в их молекулах карбоксильной группы –СООН, так и основные свойства, обусловленные аминогруппой – NH2.
И в растворе и в кристаллическом состоянии аминокислоты могут существовать в виде внутренних солей (биполярных ионов), которые образуются за счет того, что карбоксильная группа отдает протон, а аминогруппа его присоединяет:

1 Аминокислоты могут реагировать со спиртами в присутствии газообразного хлороводорода, превращаясь в сложный эфир (точнее, в хлороводородную соль эфира):

Реакция этерификации – реакция между спиртом и кислотой с выделением воды и образованием сложного эфира.
Сложные эфиры аминокислот не имеют биполярной структуры и являются летучими соединениями.
Эта реакция имеет внешнюю аналогию с реакцией нейтрализации кислоты щелочью, хотя сложные эфиры по свойствам нисколько не напоминают соли.
2) Реакции по карбоксильной группе
Образование солей по карбоксильной группе. Подобно карбоновым кислотам, аминокислоты образуют соли при действии оснований, активных металлов, основных оксидов и солей более слабых кислот.
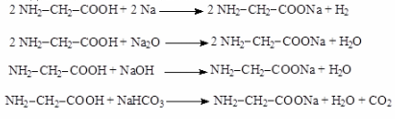
3) Образование амидов по карбоксильной группе
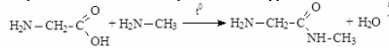
4)Реакции по аминогруппе
В растворе аминокислоты могут выступать в роли, как кислот, так и оснований, т. е. они являются амфотерными соединениями. Карбоксильная группа -СООН способна отдавать протон, функционируя как кислота, а аминная - NH2 — принимать протон, проявляя таким образом свойства основания.
Реакциями алкилирования называют реакции, включающие замену атома водорода органического соединения алкильным радикалом.
Алкилы (алкильные радикалы) — одновалентные радикалы насыщенных углеводородов (алканов), например метил - СН3 — это радикал метана CH4, этил -C2H5 — радикал этана C2H6.
Подобно аминам, аминокислоты образуют соли при действии кислот:
Н2N–CH2–COOH + HCl [H3N–CH2–COOH]+ Cl–
а) Алкилирование аминогруппы
Н2N–CH2–COOH CH3–J CH3–NH–CH2–COOH + HJ
б) Ацилирование аминогруппы (образование амидов по аминогруппе).
Реакция может протекать как под действием карбоновых кислот, так и под действием их производных – ангидридов и галогенангидридов.
H2N–CH2–COOH + CH3–COCl HN–CH2–COOH + HCl
CH3–C = O
с) Реакция с азотистой кислотой
CH3–CH–COOH + HO–N=O CH3–CH–COOH + N2 + H2O
NH2 OH
5) Специфические реакции
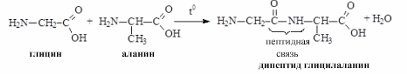
Образование пептидов. Аминогруппа одной аминокислоты может взаимодействовать с карбоксильной группой второй, образуя пептиды (соединения, состоящие из остатков аминокислот, связанных пептидной связью):
O
H2N–CH2–COOH + H2N–CH–COOH H2N–CH2–C–NH–CH–COOH + H2O
глицин аланин CH3
дипептид глицилалании
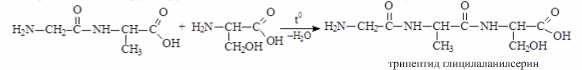
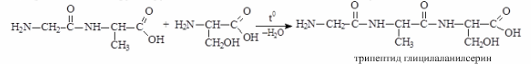
Дипептид имеет свободные –NH2 и –COOH группы и может взаимодействовать еще с одной молекулой аминокислоты, образуя трипептид, затем еще с одной молекулой – тетрапептид и т.д. При соединении друг с другом большого числа аминокислот образуются полипептиды.

дипептид глицилалании серин трипептид глицилаланинсерин
Полипептидная цепь – основа белковых молекул, белки – это природные полипептиды.
Поликонденсация. Поликонденсацией – аминокапроновой кислоты (6-аминогексановой кислоты) получают синтетическое волокно «капрон».
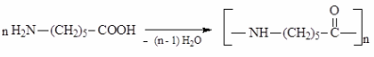

Как мы увидим далее, поликонденсация аминокислот (отличных от тех, которые образуют полипептиды и белки) используется при получении очень ценных синтетических волокон, например капрона.
СПОСОБЫ ПОЛУЧЕНИЯ АМИНОКИСЛОТ
1)Гидролиз белков:

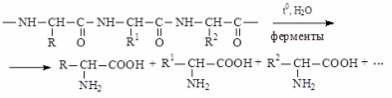
Если в недавнем прошлом -аминокислоты получали в небольших количествах, преимущественно для научных исследований, то в настоящее время налажено их многотоннажное промышленное производство. Это связано, например, с тем, что -аминокислоты являются необходимым компонентом комбикормов и синтетической пищи на углеводной основе.
2) Из галогенопроизводных карбоновых кислот
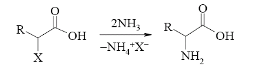 где: Х – Cl, Br
где: Х – Cl, Br
Монохлорпропионовая Аланин,
к-та -аминопропионовая кислота
CH2–COOH CH2COOH
| |
Cl NH2
Монохлоруксусная Глицин
кислота -аминоуксусная кислота
Метод применим для синтеза не только -аминокислот, но и аминокислот с любым отдалением аминогруппы от карбоксильной. Выход аминокислот составляют 70-80% при 10-12 кратном избытке аммиака и добавлении карбоната аммония.
При взаимодействии -хлор- или -бромкарбоновых кислот с избытком водного, спиртового или жидкого аммиака, в результате нуклеофильного замещения образуются соответствующие -аминокислоты.
2) В лабораторных условиях химический синтез аминокислот осуществляют при взаимодействии галогенокарбоновых кислот с аммиаком.
Исходные -галогенокислоты обычно получают по реакции Гелля–Фольгарда–Зелинского:
По этому методу из аммиака, альдегидов и синильной кислоты получают -аминонитрилы, гидролиз которых дает -аминокислоты.
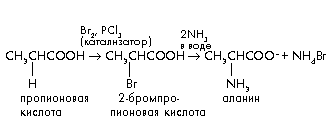
3) Синтез Штреккера
В синтезе Штреккера альдегид превращают в -аминокислоту с удлинением углеродной цепи на один атом углерода.
По этому методу из аммиака, альдегидов и синильной кислоты получают -аминонитрилы, гидролиз которых дает -аминокислоты.
Процесс проходит в две стадии.
На первой стадии в результате реакции альдегида с NH4Cl и NaCN получают -аминонитрил, на второй – при гидролизе нитрильной группы -аминонитрила получают аминокислоту.
а) получение аланина:
б) получение фенилаланина:
Основными недостатками метода являются:
- относительно низкий выход – аминокислот,
- применение токсичного цианистого водорода.
4) Очень распространен способ получения -аминокислот с использованием малонового эфира.
При этом один из атомов водорода метиленовой группы малонового эфира замещается на аминогруппу, а другой – на соответствующий требуемой аминокислоте углеводородный радикал. В результате гидролиза полученного диэтилового эфира и последующего декарбоксилирования дикислоты получают нужную аминокислоту:
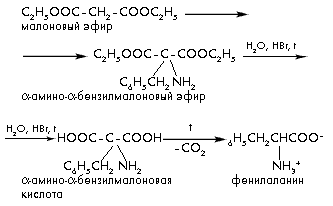
5) Еще один способ синтеза аминокислот заключается в восстановительном аминировании (восстановлении водородом в присутствии аммиака) -оксокарбоновых кислот:
(CH3)2CHCH2CCOOH + NH3 + H2 (CH3)2CH–CH2–CH–COOH
O NH2
4- метил-2-оксипентановая лейцин
кислота
ХИМИЧЕСКИЕ СВОЙСТВА АМИНОКИСЛОТ
В растворе аминокислоты могут выступать в роли как кислот, так и оснований, т. е. они являются амфотерными соединениями. Карбоксильная группа -СООН способна отдавать протон, функционируя как кислота, а аминная - NH2 — принимать протон, проявляя таким образом свойства основания.
1)Аминокислоты взаимодействуют с кислотами:
NH2 – CH2 – COOH + HCI HCI• NH2 – CH2 –COOH
(хлороводородная соль глицина)
2)Аминокислоты взаимодействуют с щелочами:
NH2 —CH2 —COOH + NaOH NH2 —CH2 —COONa + H2O
(натриевая соль глицина)
3) Дезаминирование азотистой кислотой:
H2N-CH(R)-COOH + HNO2 HO-CH(R)-COOH + N2+ H2O
Подобно первичным аминам, аминокислоты реагируют с азотистой кислотой, при этом аминогруппа превращается в гидроксогруппу, а аминокислота — в гидроксикислоту:
Измерение объема выделившегося азота позволяет определить количество аминокислоты (метод Ван-Слайка).
4) Реакция этерификации (со спиртами в присутствии газообразного хлороводорода (НСI)
H2N-CH(R)-COOH + R'OH H2N-CH(R)-COOR' + Н2О.
Аминокислоты могут реагировать со спиртами в присутствии газообразного хлороводорода, превращаясь в сложный эфир (точнее, в хлороводородную соль эфира).
Сложные эфиры аминокислот не имеют биполярной структуры и являются летучими соединениями.
Реакция этерификации – реакция между спиртом и кислотой с выделением воды и образованием сложного эфира. Эта реакция имеет внешнюю аналогию с реакцией нейтрализации кислоты щелочью, хотя сложные эфиры по свойствам нисколько не напоминают соли.
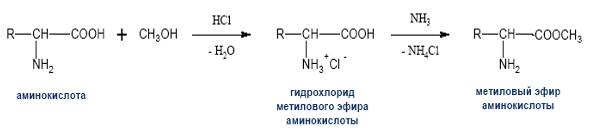
NH2–CH2– COOH+CH3OH H2O+NH2–CH2 COOCH3
(метиловый эфир глицина)
Реакция этерификации – реакция между спиртом и кислотой с выделением воды и образованием сложного эфира. Эта реакция имеет внешнюю аналогию с реакцией нейтрализации кислоты щелочью, хотя сложные эфиры по свойствам нисколько не напоминают соли.
6) Алкилирование аминогруппы:

Реакциями алкилирования называют реакции, включающие замену атома водорода органического соединения алкильным радикалом.
Алкилы (алкильные радикалы) — одновалентные радикалы насыщенных углеводородов (алканов), например метил -СН3 — это радикал метана CH4, этил -C2H5 — радикал этана C2H6.
7) Превращение аминокислот в различных средах
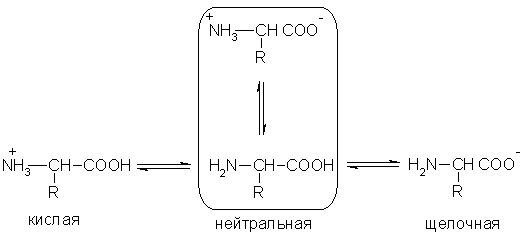
1) Аминокислоты - это органические амфотерные соединения. Они содержат в составе молекулы две функциональные группы противоположного характера: амино группу с основными свойствами и карбоксильную группу с кис лотными свойствами. Аминокислоты реагируют как с кислотами, так и с основаниями:
2) При растворении аминокислот в воде карбоксильная группа отщепляет ион водорода, который может присоединиться к ами ногруппе. При этом образуется внутренняя соль, молекула которой представляет собой биполярный ион:
+NH3– CH COO —
|
R
3) Кислотно-основные превращения аминокислот в различных средах можно изобразить следующей общей схемой:
4) Водные растворы аминокислот имеют нейтральную, щелочную или кислую среду в зависимости от количества функциональных групп.
Так, глутаминовая кислота образует кислый раствор (две группы -СООН, одна -NH2), лизин - щелочной (одна группа -СООН, две -NH2).
8) Важнейшее свойство аминокислот — их способность к конденсации с образованием пептидов.
Аминогруппа одной аминокислоты может взаимодействовать с карбоксильной группой второй, образуя пептиды (соединения, состоящие из остатков аминокислот, связанных пептидной связью):
Дипептид имеет свободные –NH2 и –COOH группы и может взаимодействовать еще с одной молекулой аминогруппы, образуя трипептид, затем еще с одной молекулой – тетрапептид и т.д. При соединении друг с другом большого числа аминокислот образуются полипептиды.
Полипептидная цепь – основа белковых молекул, белки – это природные полипептиды.
БИОЛОГИЧЕСКИЙ СИНТЕЗ АМИНОКИСЛОТ
В живых организмах аминокислоты синтезируются без участия неорганических катализаторов и высоких температур с помощью ферментативных процессов включения аммиака в органические соединения.
Известны три основные реакции такого включения:
- аминирование,
- переаминирование и
- включение аммиака в пиримидины и мочевину.
Реакция первого типа – образование глутамата из -кетоглутарата и аммиака – катализируется ферментом глутаматдегидрогеназой.
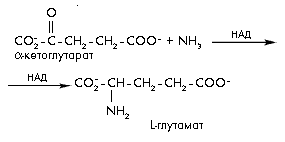
В реакциях переаминирования аминокислоты образуются из органических кислот в результате переноса аминогруппы от другой аминокислоты-донора при участии пиридоксальфосфата:
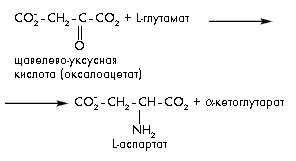
3) Наконец, большое значение имеют реакции третьего типа, приводящие к включению аммиака в мочевину и катализируемые карбамоилфосфатсинтазой:
СO2 + NH3 + 2AТФ + H2O NH2–C–OPO3H– + 2АДФ + Ф
ФИЗИЧЕСКИЕ СВОЙСТВА АМИНОКИСЛОТ
Аминокислоты — бесцветные кристаллические вещества, хорошо растворимые в воде, нерастворимые в неполярных растворителях, растворы многих аминокислот имеют сладкий вкус. Аминокислоты при нагревании разлагаются, поэтому не имеют точных температур кипения и плавления.
ПЕПТИДЫ
Аминогруппа одной аминокислоты способна вступать в реакцию с карбоксильной группой другой аминокислоты. Образующаяся при этом молекула представляет собой дипептид, а связь -CO-NH- называется пептидной связью.
На одном конце молекулы дипептида находится свободная аминогруппа, а на другом — свободная карбоксильная группа. Благодаря этому дипептид может присоединять к себе другие аминокислоты, образуя олигопептиды. Если таким образом соединяется много аминокислот (более десяти), то получается полипептид.
Пептиды играют важную роль в организме. Многие олиго- и полипептиды являются гормонами, антибиотиками, токсинами.
К олигопептидам относятся окситоцин, вазопрессин, тиреотропин, а также брадикинин (пептид боли) и некоторые опиаты («естественные наркотики» человека), выполняющие функцию обезболивания. Принятие наркотиков разрушает опиатную систему организма, поэтому наркоман без дозы наркотиков испытывает сильную боль — «ломку», которая в норме снимается опиатами. К олигопептидам относятся и некоторые антибиотики (например, грамицидин S).
Многие гормоны (инсулин, адренокортикотропный гормон и др,), антибиотики (например, грамицидин А), токсины (например, дифтерийный токсин) являются полипептидами.
Образование пептидной связи
В природе в составе белков встречается 20 аминокислот, при этом все они обычно являются левовращающими (L-изомеры), т.е. закручивают угол поляризации плоскополяризованного света влево при прохождении им раствора АК.
Постройка полипептидной (белковой) цепи происходит путем образования между молекулами АК пептидных связей. Белки, в зависимости от последовательности АК в их составе, образуют сложные пространственные структуры, соответствующие их клеточным функциям. Для нас важно, что и в процессе репликации, и в процессе трансляции в современных организмах белки принимают непосредственное участие, реализуя свою ферментативную функцию.
Синтезировать можно многие тысячи различных аминокислот, и множество различных аминокислот встречается в природе, но для синтеза белков используется только 20 видов аминокислот: аланин, аргинин, аспарагин, аспарагиновая кислота, валин, гистидин, глицин, глутамин, глутаминовая кислота, изолейцин, лейцин, лизин, метионин, пролин, серин, тирозин, треонин, триптофан, фенилаланин и цистеин (в белках цистеин может присутствовать в виде димера - цистина). Правда, в некоторых белках присутствуют и другие аминокислоты, помимо регулярно встречающихся двадцати, но они образуются в результате модификации какой-нибудь из двадцати перечисленных уже после того, как она включилась в белок.
КАЧЕСТВЕННЫЕ РЕАКЦИИ НА АМИНОКИСЛОТЫ
1) Биуретовая реакция
При взаимодействии 1-2 мл разбавленного белка с 2-3 мл 1% раствора CuSO4 в щелочной среде (2-4 мл 30% раствора NaOH) развивается фиолетовое окрашивание.
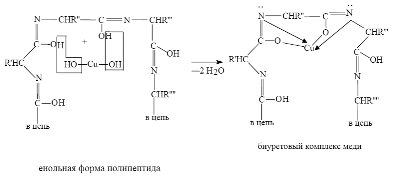
Механизм данной реакции связан с образованием комплексных соединений меди с атомами азота полипептидной цепи. Щелочная среда требуется для депротонирования атомов азота.
2) Нингидриновая реакция на -аминокислоты
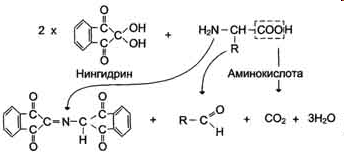
Эта реакция основана на том, что бесцветный нингидрин, реагируя с аминокислотой, конденсируется в виде димера через атом азота, отщепляемый от -аминогруппы аминокислоты. В результате образуется пигмент красно-фиолетового цвета. Одновременно происходит декарбоксилирование аминокислоты, что приводит к образованию СО2 и соответствующего альдегида. Нингидриновую реакцию широко используют при изучении первичной структуры белков (см. схему ниже).
Аргинин определяют с помощью качественной реакции на гуанидиновую группу (реакция Сакагучи), а цистеин выявляют реакцией Фоля, специфичной на SH-группу данной аминокислоты. Наличие ароматических аминокислот в растворе определяют ксантопротеиновой реакцией (реакция нитрования), а наличие гидроксильной группы в ароматическом кольце тирозина - с помощью реакции Миллона.
При взаимодействии белка со спиртовым раствором нингидринапри температуре 700 С развивается сине-фиолетовое окрашивание.
Реакция проходит в 2 стадии:
1- стадия. Эта реакция основана на том, что бесцветный нингидрин, реагируя с аминокислотой с образованием дикетодигринамина. Одновременно происходит декарбоксилирование аминокислоты, что приводит к образованию СО2 и соответствующего альдегида.
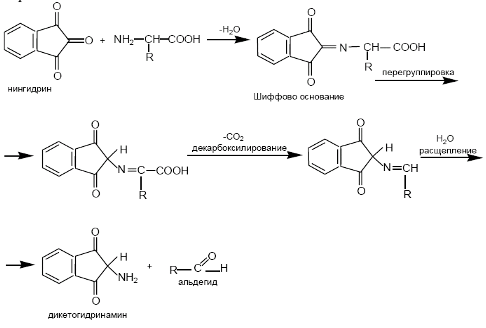
2 стадия. Дикетодигринамин конденсируется в виде димера через атом азота, отщепляемый от -аминогруппы аминокислоты.
В результате образуется пигмент сине-фиолетового цвета (пурпура Руэманна).
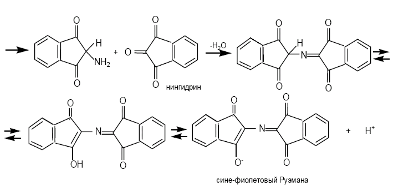
Нингидриновую реакцию широко используют при изучении первичной структуры белков (см. схему ниже).
3) Ксантопротеиновая реакция – например, по ароматическому кольцу тирозина
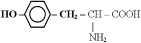
При взаимодействии белка с концентрированной азотной кислотой белок выпадает в осадок. При нагревании раствор и осадок окрашиваются в ярко-желтый цвет. При этом осадок почти полностью растворяется.
Смесь охладить и по каплям добавить избыток NH4OH или NaOH до щелочной реакции. Вначале выпадает осадок кислотного альбумина растворяется и раствор окрашивается в ярко-оранжевый цвет.
Переход желтой окраски в оранжевую в щелочной среде обусловлен изменением структуры щелочных солей этих нитросоединений.
Желатина, не содержащая ароматических аминокислот, не дает этой реакции.
2) 3)
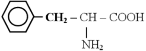
фенилаланин триптофан
Ксантопротеиновая реакция зависит от наличия в белках остатков ароматических аминокислот [фенилаланина (2), тирозина (1), триптофана (3)].
Эти аминокислоты в результате нитрования образуют желтоокрашенные нитросоединения.
4) реакция с азотно-ртутным реактивом Миллона
При взаимодействии белка с азотно-ртутным реактивом Миллона , белок свертывается под действием солей ртути и азотной кислоты образуя сгусток белого цвета. При нагревании на водяной бане осадок окрашивается в кирпично-красный цвет.
Реакцию Миллона дают все белки, содержащие в своем составе остаток тирозина.
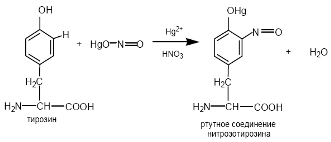
Белки, не содержащие тирозин (желатина) не дают реакцию Миллона
5) реакция Адамкевича на триптофан
При взаимодействии белка с ледяной уксусной кислотой при нагревании до растворения осадка в среде концентрированной сернокислотной кислоты образуется красно-фиолетовое окрашивание.
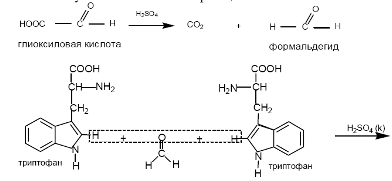
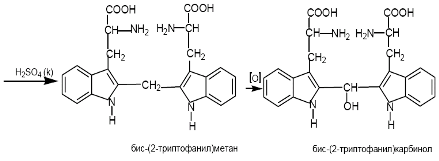
Желатина не дает этой реакции, т.к. не содержит триптофан.
Окраска возникает за счет реакции триптофана с глиоксиловой кислотой, всегда присутствующей в уксусной кислоте. Небольшое количество меди повышает чувствительность этой реакции.
6) Реакция Сакагучи на аргинин
Аргинин, содержащий гуанидиновую группировку, окисляется гипобромитом. Окисленная форма аргинина при взаимодействии с a-нафтолом образует соединение красного цвета.
7)Реакция Фоля на серосодержащие аминокислоты
Известны 3 серосодержащие аминокислоты: цистеин, цистин и метионин.
В молекулах цистеина и цистина сера связана относительно слабо и легко отщепляется при щелочном гидролизе в виде сероводорода, который реагирует со щелочью, образуя сульфиды натрия или калия.
Последние взаимодействуют с уксуснокисльм свинцом с образованием осадка сернистого свинца черного или буро-черного цвета. Реакция протекает по следующим уравнениям:

Na2S + Pb(ONa)2 + 2H2O PbS + 4NaOH
Аминокислоты табака сами по себе биологической опасности не представляют, но при сгорании табака,то есть при курении, происходит окисление аминокислот с образованием различных оксидов азота, из которых NO, NO2, N2O5 – относятся к токикантам 2 класса опасности. Высший оксид - N2O5 соединяясь в легочных альвеолах с водой образует азотную кислоту.
МЕТАБОЛИЗМ АМИНОКИСЛОТ
Продукты гидролиза белков всасываются в кишечнике в основном виде свободных аминокислот и отчасти ди- и трипептидов.
Аминокислоты в первую очередь используются в качестве строительного материала для синтеза специфических тканевых белков, ферментов, гормонов и др. биологически активных соединений. Некоторое количество аминокислот подвергается распаду с образованием конечных продуктов белкового обмена (СО2; Н2О;NH3) и освобождение энергии.
Промежуточный метаболизм аминокислот белковых молекул, как и других питательных веществ в живых организмах, включает:
- катаболические (распад до конечных продуктов обмена),
- анаболические (синтез более сложных веществ) процессы.
Условно промежуточный метаболизм аминокислот можно разделить на:
- общие пути обмена и
- индивидуальные превращения отдельных аминокислот.
Общие пути обмена аминокислот включают реакции:
- дезаминирования,
- декарбоксилирования,
- трансаминирования.
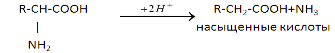
- Дезаминирование аминокислот – процесс отщепления аммиака от аминокислоты.
Существуют следующие типы дезаминирования:
1) Восстановительное
2)Гидролитическое
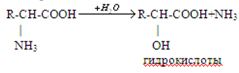
3) Внутримолекулярное - Для животных тканей, растений и большинства аэробных микроорганизмов преобладающим типам реакций является окислительное дезаминирование аминокислот, за исключением гистидина, подвергающеюся внутримолекулярному дезаминированию.
4) Окислительное - дезаминирование протекает в две стадии. Первая стадия является ферментативной и завершается образованием неустойчивого промежуточного продукта (аминокислота), который на второй стадии без участия фермента, но в присутствии воды распадается на аммиак и кетокислоту.
R–CH–COOH R– C–COOH + NH3
NH2 O
- Декарбоксилирование аминокислот - процесс отщепления карбоксильной группы аминокислоты в виде СО2.
Реакции декарбоксилирования в отличие от других процессов промежуточного обмена аминокислот являются необратимыми. Они катализируются специфическими ферментами – декарбоксилазами.
Образующиеся продукты реакции, названные биогенными аминами, оказывают сильное фармакологическое действие на множество физиологических функций человека и животных.
В животных тканях с высокой скоростью протекает декарбоксилирование гистидина с образованием биогенного амина – гистамина.
Гистамин обладает широким спектром биологического действия:
- сокращает гладкие мышцы легких;
- оказывает сосудорасширяющее действие;
- участвует в секреции соляной кислоты;
- понижает давление;
- выполняет роль медиатора боли;
- участвует в патогенезе аллергий.
Выраженное фармакологическое действие оказывают продукты декарбоксилирование ароматических кислот, глютаминовой кислоты.
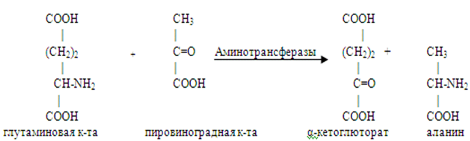
Трансаминирование аминокислот – процесс межмолекулярного переноса аминогруппы (NH2–) от аминокислоты на -кетокислоту без промежуточного образования аммиака.
Впервые реакции трансаминирования (прежнее название «переаминирование») были открыты в 1937 г. советскими учеными А.Е. Браунштейном и М.Г. Крицман при изучении дезаминирования глутаминовой кислоты в мышечной ткани.
Реакция трансаминирования являются обратимымия и универсальными для всех живых организмов.
Эти реакции протекают при участии специфических ферментов – аминотрансфераз или трансаминаз. В переносе аминогруппы участвует кофермент пиридоксальфосфат (коферментная форма витамина В6).
В тканях животных и микроорганизмов доказано существование реакций трансаминирования между монокарбоновыми амино-и кетокислотами, что можно представить в виде схемы.
Ферменты трансаминирования катализируют сначала перенос NH2 – группы на кофермент – пиродоксальфосфат, образуется промежуточное соединение – Шиффово основание, которое подвергается внутримолекулярным превращениям, что приводит к освобождению -кетокислоты и пиродоксаминфосфата. Последний на второй стадии реакции реагирует с любой другой -кетокислотой, и через стадии образования промежуточных соединений (идущих в обратном направлении) синтезируется новая аминокислота и освобождается пиридоксальфосфат.
Было замечено, что при добавлении к гомогенату мышц глутаминовой и пировиноградной кислот образуются -кетоглутаровая кислота и аланин без промежуточного свободного аммиака; добавление аланина и -кетоглутаровой кислоты приводило к образованию соответственно пировиноградной и глутаминовой кислот.
Лекция 3
БЕЛКИ
Белки также называют протеинами (от греч. «протоc» - первый, важный). Белок можно рассматривать как сложный полимер аминокислот. Число остатков -аминокислот в молекуле белка очень сильно колеблется и иногда достигает нескольких тысяч.
В природе в составе белков встречается 20 аминокислот [плюс 2 аминокислоты – Цистеин ® (cеленоцистеин - 21 аминокислота), лизин - пирролизин-22 аминокислота] , при этом все они обычно являются левовращающими (L-изомеры), т.е. закручивают угол поляризации плоскополяризованного света влево при прохождении им раствора АК.
Белки представляют собой полипептиды, в молекулу которых входит от 50 до нескольких тысяч аминокислот с относительной молекулярной массой свыше 10 000.
Каждый белок обладает своей, присущей ему последовательностью расположения аминокислотных остатков, эта последовательность определяется генетическим кодом.
Белки входят в состав всех живых организмов, но особо важную роль они играют в животных организмах, которые состоят из тех или иных форм белков (мышцы, покровные ткани, внутренние органы, хрящи, кровь).
Растения синтезируют белки (и их составные части - аминокислоты) из углекислого газа СО2 и воды Н2О за счет фотосинтеза, усваивая остальные элементы белков (азот N, фосфор Р, серу S, железо Fe, магний Mg) из растворимых солей, находящихся в почве.
Белки, поступающие в организм с животной и растительной пищей, гидролизуются, в конечном счете, до - аминокислот.
Наш организм устроен так, что часть - аминокислот незаменимые аминокислоты -должна обязательно содержаться в пище. Для взрослого человека их всего 8, для детей 10. А вот остальные заменимые аминокислоты организм синтезирует сам - был бы в достатке азот, без которого ни один белок не может существовать. Этот процесс осуществляется в печени.
Белки выполняют функцию биокатализаторов-ферментов, регулирующих скорость и направление химических реакций в организме. В комплексе с нуклеиновыми кислотами обеспечивают функции роста и передачи наследственных признаков, являются структурной основой мышц и осуществляют мышечное сокращение.
Из всего многообразия существующих аминокислот (теоретически количество возможных аминокислот неограниченно) в образовании белков участвуют только такие, у которых между аминогруппой и карбоксильной группой – всего один углеродный атом.
Пептиды
Аминогруппа одной аминокислоты способна вступать в реакцию с карбоксильной группой другой аминокислоты. Образующаяся при этом молекула представляет собой дипептид, а связь -CO-NH- называется пептидной связью.
На одном конце молекулы дипептида находится свободная аминогруппа, а на другом — свободная карбоксильная группа. Благодаря этому дипептид может присоединять к себе другие аминокислоты, образуя олигопептиды. Если таким образом соединяется много аминокислот (более десяти), то получается полипептид.
Пептиды играют важную роль в организме. Многие олиго- и полипептиды являются гормонами, антибиотиками, токсинами.
К олигопептидам относятся окситоцин, вазопрессин, тиреотропин, а также брадикинин (пептид боли) и некоторые опиаты («естественные наркотики» человека), выполняющие функцию обезболивания. Принятие наркотиков разрушает опиатную систему организма, поэтому наркоман без дозы наркотиков испытывает сильную боль — «ломку», которая в норме снимается опиатами. К олигопептидам относятся и некоторые антибиотики (например, грамицидин S).
Многие гормоны (инсулин, адренокортикотропный гормон и др,), антибиотики (например, грамицидин А), токсины (например, дифтерийный токсин) являются полипептидами.
Постройка полипептидной (белковой) цепи происходит путем образования между молекулами АК пептидных связей. Белки, в зависимости от последовательности АК в их составе, образуют сложные пространственные структуры, соответствующие их клеточным функциям. Для нас важно, что и в процессе репликации, и в процессе трансляции в современных организмах белки принимают непосредственное участие, реализуя свою ферментативную функцию.
Белки, благодаря присутствию в их составе ионных и полярных группировок (–NH2; –COOH; –SH; –OH и т.д.) существуют в водных растворах в виде заряженных частиц.
В зависимости от соотношения в белке основных (NH-аминных) и кислых (–СООН карбоксильных) группировок и рН среды молекула белка в водном растворе приобретает положительный или отрицательный заряд.
Число ионизированных групп в белке может быть увеличено или уменьшено при изменении рН среды. В кислой среде подавляется диссоциация карбоксильных групп и отрицательный суммарный заряд белка уменьшается. Наоборот, в щелочной среде подавляется ионизация аминных групп и положительный суммарный заряд белка уменьшается.
При определенном значении рН число положительных зарядов на поверхности белковой молекулы будет равным числу отрицательных зарядов и, в целом заряд молекулы белка станет равным нулю.
Добавляя к раствору белка определенное количество кислоты или щелочи, можно изменить его заряд. При определенном значении pH наступает такое состояние, при котором заряд белка становится нейтральным.
Состояние белка, при котором суммарный заряд его равен нулю, называется изоэлектрическим состоянием.
Концентрация водородных ионов (рН), при которой белок находится в изоэлектрическом состоянии называется изоэлектрической точкой белка и обозначается рI. При значении концентрация водородных ионов, равном изоэлектрической точке, аминокислоты не перемещаются в электрическом поле.
Если концентрация водородных ионов ниже изоэлектрической точки, катион аминокислоты движется к катоду, а при pH выше ИЭТ анион аминокислоты — к аноду. В изоэлектрической точке ионы белка не переносятся ни к аноду, ни к катоду. В этой точке достигают своего минимального значения такие свойства белков как: набухание, вязкость, электропроводность. Резко падает растворимость белка и увеличивается его способность к свертыванию.
На этих свойствах аминокислот основана возможность разделения их в электрическом поле (электрофорез).
Кислые аминокислоты имеют ИЭТ в слабокислой среде, основные — в слабоосновной, а нейтральные — в нейтральной.
В изоэлектрической точке отсутствие заряда у молекул белка ослабляет силы отталкивания между белковыми частицами, что благоприятствует агрегации белковых молекул и выпадению белка в осадок, т.е. в изоэлектрической точке раствор белка неустойчив, так как белок теряет один из факторов стабилизации белковых водных растворов – заряд.
При добавлении щелочи или кислоты к белку, выпавшему в осадок в изоэлектрическом состоянии, наступает перезарядка его молекул, и белок вновь переходит в раствор – растворяется.
Так например изоэлектрическая точка (рI) белков … лежит при следующих рН:
Желатин -4,2; Казеин – 4,6; Альбумин яйца – 4,8; Альбумин сыворотки крови -4,8; Гемоглобин – 6,8; Гистон зобной железы 8,7.
В зависимости от соотношения в белке основных (NH-аминных) и кислых (–СООН карбоксильных) группировок и рН среды молекула белка в водном растворе приобретает положительный или отрицательный заряд.
Большинство белков животного происхождения содержат в своем составе больше дикарбоновых аминокислот аспарагиновой и глютаминовой и поэтому в водных растворах они заряжаются отрицательно (белки-анионы).
Некоторые белки содержат в своем составе значительные количества диаминокислот (аргинина, лизина, гистидина) и поэтому заряжаются положительно (белки-катионы).
Одноименный заряд молекул способствует взаимному отталкиванию частиц, что обеспечивает устойчивость их в водном растворе.
Образование пептида из двух аминокислот идет с выделением молекулы воды.
Соответственно, при образовании трипептида будет выделяться уже две молекулы воды.
При образовании белка из n – молекул аминокислот, выделится (n – 1) молекул воды. Поэтому процесс образования пептидов и белков называется не полимеризацией, а поликонденсацией.
Остаток аминокислоты со свободной – аминогруппой называется N-концевым, остаток аминокислоты с -карбоксильной группой называется С - концевым, т.е. остаток треонина в данном трипептиде будет являться N – концом, а остаток тирозина С – концом.
Последовательность соединения остатков аминокислот может быть выражена следующим образом:
Н2N-Тhr-Phe-Tyr-COOH
Название пептидов строятся, перечисляя все аминокислоты кроме С - концевой в виде радикалов, оставляя название С – конца неизменным. Трипептид Nhr-Phe-Tyr будет называться следующим образом:
треонил – фенилаланил - тирозин
Отличие пептидов от белков состоит в том, что пептиды обладают низкой молекулярной массой, а белки – высокой. Граница между ними очень размыта и лежит в районе 10000 Д (иногда приводятся значения от 5000, 6000). Важным отличием пептидов от белков является их способность проникать через полупроницаемую мембрану, то есть диализуемость.
МОЛЕКУЛА ИНСУЛИНА, построенная из 51 аминокислотного остатка, фрагменты одинаковых аминокислот отмечены соответствующей окраской фона. Содержащиеся в цепи остатки аминокислоты цистеина (сокращенное обозначение ЦИС) образуют дисульфидные мостики –S-S-, которые связывают две полимерных молекулы, либо образуют перемычки внутри одной цепи.
Молекулы аминокислоты цистеина HS – CH2 – CH – COOH
NH2
содержат реакционно-способные сульфгидридные группы –SH, которые взаимодействуют между собой, образуя дисульфидные мостики –S-S-.
Роль цистеина в мире белков особая, с его участием образуются поперечные сшивки между полимерными белковыми молекулами.
Объединение аминокислот в полимерную цепь происходит в живом организме под управлением нуклеиновых кислот, именно они обеспечивают строгий порядок сборки и регулируют фиксированную длину полимерной молекулы
Шесть молекул инсулина ассоциированы в гексамер (видны три симметричные оси). Молекулы удерживают вместе остатки гистидина, связанные ионами цинка. Введенный инсулин находится под кожей в виде гексамера, постепенно распадаясь на биологически активные мономеры, поступающие в кровоток.

Рисунок – Ассоциация 6 молекул инсулина
КЛАССИФИКАЦИЯ ПЕПТИДОВ
1 По числу аминокислотных остатков:
дипептиды, трипептиды, тетрапептиды, пентапептиды;
2 По наличию циклов: ациклические и циклические;
3 По связям между остатками аминокислот: гомодетные пептиды, гетеродетные пептиды;
4 Классификация может сочетаться:
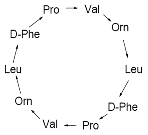
4 – например, антибиотик – грамицидин S – это циклический, гомодетный декапептид (т.е. он содержит 10 аминокислотных остатков, которые связаны исключительно пептидными связями, образуя цикл).
ФУНКЦИИ ПЕПТИДОВ
Основная функция пептидов:
1) Регулирующая функция:
гормоны (окситоцин, вазопрессин, инсулин, глюкагон, соматостатин, эндорфины);
2) Бактериостатический эффект: антибиотик (грамицидин, аманитин – пептид бледной поганки)
УРОВНИ СТРУКТУРНОЙ ОРГАНИЗАЦИИ БЕЛКОВ
Как уже было сказано, белки представляют собой полипептиды, в молекулу которых входит от пятидесяти до нескольких тысяч аминокислот с относительной молекулярной массой свыше 10 000.
СТРУКТУРА БЕЛКОВ
Какова же структура белка?
Каждому белку в определенной среде свойственна особая пространственная структура. При характеристике пространственной (трехмерной) структуры выделяют четыре уровня организации молекул белков.
Первичная структура — последовательность аминокислот в полипептидной цепи. Такая структура специфична для каждого белка и определяется генетической информацией, т. е. зависит от последовательности нуклеотидов в участке молекулы ДНК, кодирующем данный белок. От первичной структуры зависят все свойства и функции белков. Замена одной-единственной аминокислоты в составе молекул белка или нарушение порядка в их расположении обычно влечет за собой изменение функции белка.
Постулаты (принципы формирования пептидной связи), сформулированные Л. Поллингом и Р. Кори:
1) атомы, образующие пептидную связь, копланарны (расположены в одной плоскости); вращение атомов или групп атомов вокруг пептидной связи невозможно;
2) принцип эквивалентности вклада АК-остатков в образование пептидной связи и, тем самым, в образование полипептидной цепи (исключение пролин);
3) принцип максимума водородных связей.
Первичную структуру белка стабилизируют (поддерживают):
пептидные связи (между АК-остатками);
дисульфидные связи (между свободными –SH-группами цистеина).
Первичная структура белка несет информацию о его пространственной структуре.
Первичная структура — ковалентно связанные мономеры в полимер.
|
Лиз-глу-тре-ала-ала-ала-лиз-фен-глу-арг-глн-гиc-мет-асп-сер-сер-тре-сер-ала-ала-сер-сер сер-асн-тир-цис-асн-глу-мет-мет-лиз-сер-арг-асн-лей-тре-лиз-асп-арг-цис-лиз-про-вал-асн-тре-фен-вал-гис-глу-сер-лей-ала-асп-вал-глн-ала-вал-цис-сер-глн-лиз-асн-вал-ала-цис-лиз-асн-гли-глн-тре-асн-цис-три-глн-сер-три-сер-тре-мет-сер-иле-тре-асп-цис-арг-глу-тре-гли-сер-сер-лиэ-тир-про-асн-цис-ала-тир-лиэ- тре-тре-глн-ала-асн-лиз-гис-иле-иле-вал-ала-цис-глу-гли-асн-про-тир-вал-про-вал-гис-фен-асп-ала-сер-вал |
Рисунок - Аминокислотная последовательность белка рибонуклеазы (124 аминокислотных звена)
В живых клетках молекулы белков или отдельные их участки представляют собой не вытянутую цепь, а скручены в спираль, напоминающую растянутую пружину (это так называемая а-спираль), или сложены в складчатый слой (р-слой).
Такие -спирали и р-слои являются вторичной структурой.
Вторичная структура белка — локальная конформация, обусловленная вращением отдельных участков полипептидной цепи вокруг одинарных ковалентных связей. Она возникает в результате образования водородных связей внутри одной полипептидной цепи (спиральная конфигурация) или между двумя полипептидными цепями (складчатые слои).
Вторичная структура—полипептидная цепь закручена в виде спирали, локальные упорядоченные структуры полимера - полностью -спиральную конфигурацию имеет белок кератин. Это структурный белок волос, ногтей, когтей, клюва, перьев и рогов; он входит в состав наружного слоя кожи позвоночных.
Третичная структура белка — это расположение в пространстве всей полипептидной цепи, отдельные участки которой имеют собственную локальную конформацию.
Третичная структура белка –-миоглобина - полная укладка в пространстве одной цепи полимера .
Поддержанию третичной структуры белка способствуют гидрофобные связи, которые образуются внутри молекулы. В образовании этих связей принимают участие неполярные радикалы аминокислот. Могут также образовываться другие нековалентные связи.
У большинства белков спиральные и неспиральные участки полипептидной цепи складываются в трехмерное образование шаровидной формы — глобулу (характерна для глобулярных белков). Глобула определенной конфигурации является третичной структурой белка. Такая структура стабилизируется ионными, водородными, ковалентными дисульфидными связями (образуются между атомами серы, входящими в состав цистеина, цистина и мегионина), а также гидрофобными взаимодействиями. Наиболее важными в возникновении третичной структуры являются гидрофобные взаимодействия; белок при этом свертывается таким образом, что его гидрофобные боковые цепи скрыты внутри молекулы, т. е. защищены от соприкосновения с водой, а гидрофильные боковые цепи, наоборот, выставлены наружу.
Четвертичная структура формируется при объединении нескольких полипептидных цепей, имеющих третичную структуру. Образованный таким образом белок обладает новой функцией.
Четвертичная структура — гемоглобина – укладка субъединиц в пространстве.
Белки с четвертичной структурой называются олигомерными, а составляющие их индивидуальные полипептидные цепи — протомерами или мономерами. Такие соединения стабилизируются водородными связями и электростатическими взаимодействиями между АК-остатками, расположенными на поверхности протомеров.
Преимущества белков с четвертичной структурой:
1) экономия генетического материала;
2) уменьшение числа ошибок при синтезе белка;
3) качественное разнообразие белков — появление у белков новых функций.
Многие белки с особо сложным строением состоят из нескольких полипептидных цепей (субъединиц), образуя четвертичную структуру белковой молекулы. Такая структура имеется, например, у глобулярного белка гемоглобина. Его молекула состоит из четырех отдельных полипептидных субъединиц (протомеров), находящихся в третичной структуре, и небелковой части — гема.
Только в такой структуре гемоглобин способен выполнять свою транспортную функцию.
Примером различной структуры белка может служить молекула инсулина.
А - Инсулин: первичная структура
Б - Вторичная структура
Вторичными структурами называются участки полипептидной цепи с упорядоченной конформацией, стабилизированной водородными связями. В большинстве глобулярных белков присутствуют одновременно как -спирали, так и -складчатые листы. Кроме того, имеются участки с неупорядоченной структурой. Распространенным структурным элементом глобулярных белков является -петля.
В молекуле инсулина участки, имеющие форму -спирали, составляют 57%, 6% приходится на -складчатую структуру, 10% построено в виде -петли, оставшиеся 27% не имеют упорядоченной структуры.
В - Третичная структура
Трехмерные функционально активные конформации белков носят название третичной структуры. Третичную структуру белков исследуют главным образом методом кристаллографии. Этот трудоемкий метод основан на дифракции рентгеновских лучей на хорошо сформированных белковых кристаллах. Ha основании дифракционных картин рассчитывают распределение электронной плотности в кристалле, а по электронной плотности восстанавливают пространственную структуру молекул белка с атомным разрешением. В настоящее время определены трехмерные структуры сотен белков. Однако многие белки пока нельзя изучить этим методом, поскольку их не удается получить и виде хорошо сформированных кристаллов достаточно крупных размеров.
Анализ третичной структуры инсулина показал, что в -цепи имеются два коротких участка, а в В-цепи — один длинный участок, построенные в виде -спирали (1). При этом N-конец А-цепи и С-конец В-цепи располагаются в непосредственной близости друг от друга. Единственная структура типа складчатого листа образуется в димере инсулина (см. Г, 4). Третичная структура проинсулина еще не установлена.
Г - Четвертичная структура
Белковые молекулы часто образуют симметрично построенные комплексы, стабилизированные за счет нековалентныч взаимодействий. Такие комплексы называются олигомерами, а составные единицы комплексов (от 2 до 12) - субъединицами или мономерами. Инсулин также образует четвертичные структуры. В крови инсулин присутствует частично в виде димера (1). Димер имеет ось симметрии второго порядка. Кроме того, в поджелудочной железе в качестве запасной формы содержится гексамер инсулина (из 6 мономеров), стабилизированный ионами Zn2+. В образовании двух комплексов с катионом Zn2+ принимают участие остатки гистидина в положении B-10 всех шести субъединиц. На схеме 2 показано, что каждый октаэдрический комплекс включает один катион Zn2+, три остатка гистидина и три молекулы воды.
Под влиянием различных химических и физических факторов (обработка спиртом, ацетоном, кислотами, щелочами, высокой температурой, облучением, высоким давлением и т. д.) происходит изменение вторичной, третичной и четвертичной структур белка вследствие разрыва водородных и ионных связей. Процесс нарушения нативной (естественной) структуры белка называется денатурацией. При этом наблюдается уменьшение растворимости белка, изменение формы и размеров молекул, потеря ферментативной активности и т. д. Процесс денатурации может быть полным или частичным. В некоторых случаях переход к нормальным условиям среды сопровождается самопроизвольным восстановлением естественной структуры белка. Такой процесс называется денатурацией.
ВЛИЯНИЕ ХИМИЧЕСКИХ И ФИЗИЧЕСКИХ ФАКТОРОВ НА БЕЛКИ
Денатурация белка
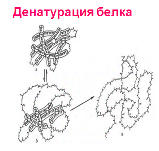
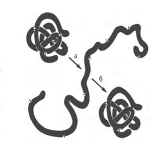
А Б
Рисунок -А - Денатурация белковой молекулы (схема)
а - исходное состояние; б - начинающееся обратимое нарушение молекулярной структуры; в - необратимое развертывание полипептидной цепи.
Рисунок- Б - Денатурация и ренатурация рибонуклеазы (по Анфинсену).
а - развертывание (мочевина + меркаптоэтанол); б - повторное свертывание.
При непродолжительном действии и быстром удалении денатурирующих агентов возможна ренатурация белка с полным восстановлением исходной трехмерной структуры и нативных свойств его молекулы (рис. 1.13), включая биологическую активность.
Таким образом, при денатурации белковая молекула полностью теряет биологические свойства, демонстрируя тем самым тесную связь между структурой и функцией.
Для практических целей иногда используют процесс денатурации в «мягких» условиях, например при получении ферментов или других биологически активных белковых препаратов в условиях низких температур в присутствии солей и при соответствующем значении рН .
При лиофилизации белков (высушивание в вакууме путем возгонки влаги из замороженного состояния) для предотвращения денатурации часто пользуются химическими веществами (простые сахара, глицерин, органические анионы).
Лекция 4
МНОГООБРАЗИЕ БЕЛКОВ
В организме человека содержится свыше 50 000 индивидуальных белков, отличающихся первичной структурой, конформацией, строением активного центра и функциями. Белки построены из 20 химически различных аминокислот, каждая из которых может занимать любое положение в полипептидной цепи. Кроме того, белки различаются количеством аминокислот, из которых они построены.
Однако большинство таких белков в среде должны принимать множество конформаций с приблизительно одинаковой энергией, но разными химическими свойствами и функциями. Поэтому в эволюции, по-видимому, была отобрана лишь небольшая часть возможных вариантов белков, которые способны принимать единственную стабильную конформацию.
Таким образом, первичная структура известных белков, отобранных эволюцией, обеспечивает исключительную стабильность одной из возможных конформаций, которая и определяет особенности функционирования данного белка.
Возникновение новых белков часто связано с незначительными изменениями в структуре уже имеющихся белков. Кроме того, благодаря генетическим механизмам, о которых будет сказано в разделе 4, белок с полезными свойствами или основная структурная часть этого белка могут входить в состав других белков. Такие белки, имеющие схожую последовательность аминокислот и родственные функции, объединяют в семейства родственных белков.
Классификация белков
До настоящего времен нет единой и стройной классификации, учитывающей различные параметры белков. В основе имеющихся классификаций обычно лежит один признак. Так, белки можно классифицировать:
- по форме молекул (глобулярные или фибриллярные);
- по молекулярной массе (низкомолекулярные, высокомолекулярные и др.);
- по химическому строению (наличие или отсутствие небелковой части);
- по выполняемым функциям (транспортные, защитные, структурные белки и др.);
- по локализации в клетке (ядерные, цито-плазматические, лизосомальные и др.);
- по локализации в организме (белки крови, печени, сердца и др.);
- по способности адаптивно регулировать синтез белков: белки, синтезирующиеся с постоянной скоростью (конститутивные), и белки, синтез которых может усиливаться при воздействии факторов среды (индуцибельные);
- по продолжительности жизни в клетке (от очень быстро обновляющихся белков, с Т1/2 менее 1 ч, до очень медленно обновляющихся белков, Т1/2 которых исчисляют неделями и месяцами);
- по схожим участкам первичной структуры и родственным функциям (семейства белков).
Классификация белков по форме молекул
Это одна из самых старых классификаций, которая делит белки на 2 группы: глобулярные и фибриллярные. К глобулярным относят белки, соотношение продольной и поперечной осей которых не превышает 1:10, а чаще составляет 1:3 или 1:4, т.е. белковая молекула имеет форму эллипса. Большинство индивидуальных белков человека относят к глобулярным белкам. Они имеют компактную структуру и многие из них, за счёт удаления гидрофобных радикалов внутрь молекулы, хорошо растворимы в воде. Наглядные примеры строения и функционирования глобулярных белков - рассмотренные выше миоглобин и гемоглобины.
Фибриллярные белки имеют вытянутую, нитевидную структуру, в которой соотношение продольной и поперечной осей составляет более 1:10. К фибриллярным белкам относят коллагены, эластин, кератин, выполняющие в организме человека структурную функцию, а также миозин, участвующий в мышечном сокращении, и фибрин - белок свёртывающей системы крови. На примере коллагенов и эластина рассмотрим особенности строения этих белков и связь их строения с функцией.
Гемоглобин состоит из белка глобина (который состоит из 18 аминокислот: аланин, валин, лейцин, пролин, цистен, цистин, аргинин и т.д. Нет – Изолейцина, оксипролина, оксилизина) и железосодержащего гема. Большая часть гемоглобина у взрослых состоит из двух альфа- и двух бета-цепей глобина (по 141 и 146 аминокислот соответственно). В каждую цепь глобина встроена молекула гема; содержащийся в ней атом железа связывает кислород. Переносить кислород может только двухвалентное железо.
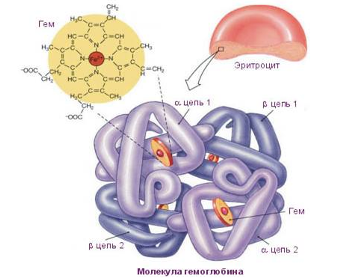
Рисунок – Молекула гемоглобима
Химически гемоглобин относится к группе хромопротеидов. Каждая молекула гемоглобина состоит из белковой части: две цепи (субъединицы) альфа-глобина и две (субъединицы) бета-глобина, нековалентно связаны, группы небелковой природы-гема, в состав которого входит железо. Каждая субъединица имеет молекулярную массу около 16000, в общей молекулярной массой в тетрамер около 64000. Каждая субъединица гемоглобина содержит одну гема , таким образом, общая связывающая способность взрослого человека гемоглобина к кислороду в четыре молекулы кислорода.
Альфа-полипептидная цепь заканчивается комбинацией аминокислот валина-лейцина, а бета-полипептидная цепь - комбинацией валина-гистидина-лейцина. Альфа- и бета-полипептидные цепи в гемоглобиновой молекуле не размещены линейно, как это выглядит на первый взгляд из данных,это первичная структура.
По причине существования интрамолекулярных сил, полипептидные цепи скручиваются в форме типичной для белков альфа-геликсовой спирали -вторичная структура. Сама альфа-геликсовая спираль на каждую альфа- и бета-полипептидную цепь огибается пространственно, образуя сплетения овоидной формы, третичная структура. Отдельные части альфа-геликсовых спиралей полипептидных цепей отмечают латинскими буквами от А до Н.
Все четыре третично изогнутые альфа- и бета-полипептидные цепи располагаются пространственно в определенном соотношении - кватернерная структура. Они связаны между собой не настоящими химическими связями, а межмолекулярными силами.
Кроме координационной связи, существующей между полипептидными цепями глобина, Fe++ атом гема располагает еще тремя координационными связями , две из которых соединены двумя азотными атомами порфиринового кольца, а третья, в среде с низким парциальным давлением кислорода ,связана с одной молекулой воды . В среде с высоким парциальным давлением кислорода (артериальная кровь), третья координационная связь соединена с одной молекулой кислорода, причем получается соединение - оксигемоглобин . Путем непрерывного превращения оксигемоглобина в редуцированный гемоглобин и обратно, осуществляется перенос кислорода из легких к тканям.
Особенно значительным отличаем гемоглобина от миоглобина является кривая насыщения кислородом, которая имеет сигмоидную форму. Значит возможность гемоглобина связывать кислород зависит от того, заключаются ли в данном тетрамере другие молекулы кислорода. Если содержатся, то последующие молекулы кислорода присоединяются легче. Таким образом, для гемоглобина свойственна кинетика кооперативного связывания, благодаря которой он объединяет максимальное количество кислорода в легких и отдает максимальное количество кислорода при тех парциальных давлениях кислорода, которые имеют место в периферических тканях.
Величина Р50 - значение парциального давления кислорода характеризует сродство гемоглобинов к кислороду. Р50 у разных организмов существенно различается, но во всех случаях оно превышает значение парциального давления кислорода в периферических тканях рассматриваемого организма.
Это показывает фетальный гемоглобин человека (НВF). Для HbA Р50=26 мм. рт. ст., а для HbF Р50=20 мм. рт. ст. Благодаря этой разнице гемоглобин F отбирает кислород у HbA, находящегося в плацентарной крови. Однако после рождения ребенка HbF утрачивает свою функцию; обладая более высоким сродством к кислороду, он высвобождает меньшее его количество в тканях.
У гемоглобина есть еще одна немаловажная функция, он ускоряет транспорт углекислого газа от тканей к легким. Гемоглобин связывает углекислый газ сразу после высвобождения кислорода; примерно 15% углекислого газа, присутствующего в крови, переносится молекулами гемоглобина. Находящаяся в эритроцитах карбоангидраза катализирует превращение поступающего из тканей углекислого газа в угольную кислоту . Угольная кислота быстро диссоциирует на бикарбонат-ион и протон, причем равновесие вдвинуто в сторону диссоциации. Для предотвращения опасного повышения кислотности крови должна существовать буферная система, способная поглощать избыток протонов. Гемоглобин связывает два протона на каждые четыре освободившиеся молекулы кислорода, определяя буферную емкость крови.
В легких идет противоположный процесс: присоединение кислорода к дезоксигемоглобину сопровождается высвобождением протонов, которые связываются с бикарбонат ионами, переводя их в угольную кислоту. Далее эффективно действующая карбоангидраза катализирует превращение угольной кислоты в углекислый газ, выдыхаемый из легких. Следовательно, связывание кислорода тесно сопряжено с выдыханием углекислого газа. Это явление называется эффектом Бора.
Железо, которое содержится в гемме, способно образовывать с молекулами кислорода легко распадающееся соединение при прохождении эритроцита чрез капилляры легких, а при прохождении через сосуды других органов - отдавать кислород и связываться с углекислотой, которую гемм затем отдает, когда эритроцит вновь попадает в капилляры легких.
У взрослых людей, гемоглобин представляет собой тетрамер, состоящий из двух альфа- и бета- субъединиц, нековалентно связаные. Субъединицы структурно похожи и примерно такого же размера. Каждая субъединица имеет молекулярную массу около 16000, в общей молекулярной массой в тетрамер около 64000. Каждая субъединица гемоглобина содержит одну гема, таким образом, что общая связывающая способность взрослого человека гемоглобина к кислороду в четыре молекулы кислорода.
Поэтапная реакция:
Hb + O2 HBO2
HBO2 + O2 Hb (O2)2
Hb (O2)2 + O2 Hb (O2)3
Hb (O2)3 + O2 Hb (O2)4
Основная информация реакция:
HB + 4O2 Hb (O2)4
Кровь, протекающая по артериям насыщена кислородом, имеет ярко-алый цвет; после поглощения кислорода тканями и связывания гемоглобина с углекислотой кровь приобретает темно-красный цвет(эта кровь протекает по венам).Помимо гемоглобина крови, у ряда животных в ритмически работающих мышцах с интенсивным обменном (мышца сердца) имеется мышечный гемоглобин (миоглобин), близкий по своему составу и свойствам к гемоглобину крови.
ХИМИЧЕСКОЕ СТРОЕНИЕ
Химически гемоглобин относится к группе хромопротеидов. Его простетическая группа представляет собой ферросоединение протопорфирина IХ, с молекулярным составом С34Н32О4N4Fe и носит название гем (рис.). Она придает соединению окраску. Белковый компонент гемоглобина называется глобином. Гемоглобиновая молекула содержит 4 гема и 1 глобин. Аминокислоты расположены в глобине в виде четырех полипептидных цепочек; две из них идентичны по структуре, и их обозначают как альфа-цепочки; две другие тоже идентичны между собой и их обозначают как бета-цепочки. Следовательно, формулу глобина можно выразить как альфа-альфа/бета-бета или альфа2бета2. альфа-полипептидная цепь состоит и 141, бета-полипептидная цепь - из 146 аминокислот.
Изолейцин - одна из незаменимых аминокислот, необходимых для синтеза гемоглобина.
В норме гемоглобин содержится в крови в виде трех физиологических соединений:
оксигемоглобин (НbО2) – гемоглобин в соединении с кислородом – находится в артериальной крови, придает ей ярко-алый цвет;
восстановленный, или дезоксигемоглобин (Hb), не содержащий кислорода, – находится в венозной крови, которая имеет более темный цвет, чем артериальная;
карбгемоглобин (НbСО2) – соединение гемоглобина с углекислым газом – содержится в венозной крови. (В этом случае СО2 присоединяется не к гему, а к NH2-группам белка глобина. СО2 могут связывать как оксигемоглобин, так и дезоксигемоглобин, но последний – в большей степени.)
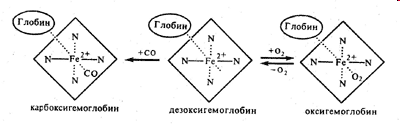
КОЛЛАГЕН - фибриллярный белок
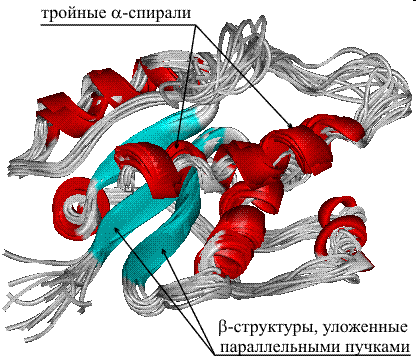
На рисунке представлена надмолекулярная структура фибриллярного белка – КОЛЛАГЕН.
Это фибриллярный белок, цепи которого построены в основном из глицина, чередующегося с пролином и лизином. Структура содержит одиночные цепи, тройные -спирали, чередующиеся с лентообразными -структурами, уложенными в виде параллельных пучков.
Их главным компонентом является фибриллярный белок коллаген (следующий слайд), наиболее распространенный белок животного мира, в организме млекопитающих, на его долю приходится почти 30% от всей массы белков.
Коллаген обладает высокой прочностью на разрыв (известна прочность кожи), но из-за малого содержания поперечных сшивок в коллагене кожи, шкуры животных мало пригодны в сыром виде для изготовления различных изделий. Чтобы уменьшить набухание кожи в воде, усадку при сушке, а также для увеличения прочности в обводненном состоянии и повышения упругости в коллагене создают дополнительные поперечные сшивки, это, так называемый процесс дубления кожи.
В живых организмах молекулы коллагена, возникшие в процессе роста и развития организма, не обновляются и не замещаются заново синтезированными. По мере старения организма увеличивается количество поперечных сшивок в коллагене, что приводит к снижению его эластичности, а поскольку обновление не происходит, то проявляются возрастные изменения – увеличение хрупкости хрящей и сухожилий, появление морщин на коже.
Строение и функции коллагенов
Коллагены - семейство родственных фибриллярных белков, секретируемых клетками соединительной ткани. Коллагены - самые распространённые белки не только межклеточного матрикса, но и организма в целом, они составляют около 1/4 всех белков организма человека. В межклеточном матриксе молекулы коллагена образуют полимеры, называемые фибриллами коллагена (более подробно это описано в разделе 15). Фибриллы коллагена обладают огромной прочностью и практически нерастяжимы. Они могут выдерживать нагрузку, в 10 000 раз превышающую их собственный вес. По прочности коллагеновые фибриллы превосходят прочность стальной проволоки того же сечения. Именно поэтому большое количество коллагеновых волокон, состоящих из коллагеновых фибрилл, входит в состав кожи, сухожилий, хрящей и костей.
Необычные механические свойства коллагенов связаны с их первичной и пространственной структурами. Молекулы коллагена состоят из трёх полипептидных цепей, называемых -цепями. Идентифицировано более 20 -цепей, большинство которых имеет в своём составе 1000 аминокислотных остатков, но цепи несколько отличаются аминокислотной последовательностью. В состав коллагенов могут входить три одинаковые или разные цепи.
Первичная структура -цепей коллагена необычна, так как каждая третья аминокислота в полипептидной цепи представлена глицином, около 1/4 аминокислотных остатков составляют пролин или 4-гидроксипролин, около 11% - аланин. В коллагене отсутствуют такие аминокислоты, как цистеин и триптофан, гистидин, метионин и тирозин находятся лишь в очень небольшом количестве. В составе первичной структуры -цепи коллагена содержится также необычная аминокислота - гидроксилизин. Полипептидную цепь коллагена можно представить как последовательность триплетов Гли-X-Y, где X и Y могут быть любыми аминокислотами, но чаще в положении X стоит пролин, а в положении Y - гидроксипролин или гидроксилизин. Каждая из этих аминокислот имеет большое значение для формирования коллагеновых фибрилл.
Пролин благодаря своей структуре вызывает изгибы в полипептидной цепи, стабилизируя ле-возакрученную спиральную конформацию. На один виток спирали приходится 3 аминокислотных остатка, а не 3,6, как это характерно для вторичной структуры глобулярных белков. Спираль пептидной цепи коллагена стабилизирована не за счёт водородных связей (так как пролин их не образует), а силами стерического отталкивания пирролидиновых колец в остатках пролина. В результате расстояние между аминокислотными остатками по оси спирали увеличивается, и она оказывается более развёрнутой по сравнению с туго закрученной -спиралью глобулярных белков.
Спирализованные полипептидные цепи, перевиваясь друг около друга, образуют трёхце-почечную правозакрученную суперспиральную молекулу, часто называемую тропоколлагеном. Цепи удерживаются друг около друга за счёт водородных связей, возникающих между амино- и карбоксильными группами пептидного остова разных полипептидных цепей, входящих в состав трёхспиральной молекулы. "Жёсткие" аминокислоты - пролин и гидроксипролин - ограничивают вращение полипептидного стержня и увеличивают тем самым стабильность тройной спирали. Глицин, имеющий вместо радикала атом водорода, всегда находится в месте пересечения цепей; отсутствие радикала позволяет цепям плотно прилегать друг к другу.
В результате такого скручивания пептидных остовов полипептидных цепей и наличия удлинённой структуры два других радикала из триады аминокислот Гли-X-Y оказываются на наружной поверхности молекулы тропоколлагена. Некоторые комплементарные участки молекул тропоколлагена могут объединяться друг с другом, формируя коллагеновые фибриллы, причём эти участки расположены таким образом, что одна нить тропоколлагена сдвинута по отношению к другой примерно на 1/4. Между радикалами аминокислот возникают ионные, водородные и гидрофобные связи.
Фибриллярные имеют вытянутую, нитевидную форму и состоят из нескольких полипептидных цепей.
Фибриллярные белки – структурные белки соединительной и покровной ткани – коллаген, эластин, кератин волос, фиброин шелка и т.д.
Фибриллярные белки устойчивы к действию кислот, щелочей, нагревания, действия протеолитических ферментов. Придают тканям и структурам плотности, жесткости, эластичности. Фибриллярные белки содержатся как внутри клеток (миофибриллы, кератины), так и в межклеточном пространстве (коллагеновые волокна).
Характерной особенностью фибриллярных белков является то, что полипептидные цепи их размещаются параллельно относительно одной оси, образуя долгие волокна.
Коллаген представляет собой белок, относящийся к классу фибриллярных. Именно фибриллярные белки отвечают за построение скелета, как всего человеческого организма, так и его отдельных кирпичиков – клеток. Изрядное количество фибриллярных белков принимает участие в формировании клеточного скелета, носящего название цитоскелет.
В переводе термин «коллаген» означает «образующий клей». Во всех без исключения многоклеточных организмах коллаген является самым распространенным белком. Коллаген – это основной компонент соединительной ткани, входящей в состав стенок кровеносных сосудов, зубов, сухожилий, костей и кожи.
Строго говоря, под коллагеном понимают не один какой-то белок, а целое семейство белков, кодируемое, как минимум, десятью структурными генами. Все эти коллагены отличаются один от другого не только пространственным строением, но и последовательностью образующих их аминокислот.
Самую большую роль в организме млекопитающих играет коллаген типа I. Это именно он входит в состав соединительной ткани кожи, сухожилий и костей.
Коллагеновые волокна соединительной ткани образованы молекулами тропоколлагена, идущими параллельно друг другу, с небольшим сдвигом. Молекула тропоколлагена представляет собой длинный стержень, в состав которого входят сразу три параллельные полипептидные цепи, закрученные в спираль. В коллагеновом волокне расположенные рядом молекулы тропоколлагена связываются друг с другом при помощи поперечных сшивок.
Любопытно, что в состав коллагена, а так же еще одного фибриллярного белка – эластина, входят аминокислоты, которые не встречаются ни в одном другом белке человеческого организма. Одной из таких аминокислот является оксипролин, образующийся в результате котрансляционной модификации пролина.
Между глобулярными и фибриллярными белками при определенных условиях возможны взаимные переходы, например, денатурация.
Строение коллагеновой фибриллы (фрагмент)
|
|
|
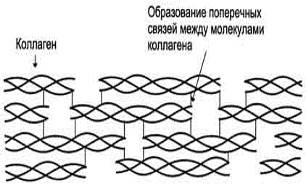
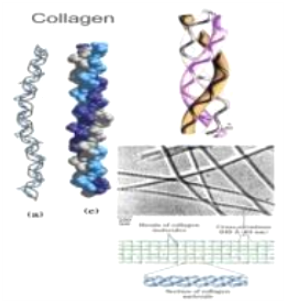
Важную роль в формировании коллагеновых фибрилл играют модифицированные аминокислоты: гидроксипролин и гидроксилизин.
Гидроксильные группы гидроксипролина соседних цепей тропоколлагена образуют водородные связи, укрепляющие структуру коллагеновых фибрилл. Радикалы лизина и гидроксилизина необходимы для образования прочных поперечных сшивок между молекулами тропоколлагена, ещё сильнее укрепляющие структуру коллагеновых фибрилл. Кроме того, к гидроксильной группе гидроксилизина могут присоединяться углеводные остатки (гликозилирование коллагена), функция которых пока неясна.
Таким образом, аминокислотная последовательность полипептидных цепей коллагена позволяет сформировать уникальную по своим механическим свойствам структуру, обладающую огромной прочностью. Изменение в первичной структуре коллагена может приводить к развитию наследственных болезней.
Для необратимого изменения формы кератинового объекта нужно вначале разрушить дисульфидные мостики с помощью восстановителя, придать новую форму, а затем вновь создать дисульфидные мостики с помощью окислителя (рис. 16), именно так делается, например, химическая завивка волос.
При увеличении содержания остатков цистеина в кератине и, соответственно, возрастании количества дисульфидных мостиков способность к деформации исчезает, но при этом появляется высокая прочность (в рогах копытных животных и панцирях черепах содержится до 18% цистеиновых фрагментов). В организме млекопитающих содержится до 30 различных типов кератина.
Родственный кератину фибриллярный белок фиброин, выделяемый гусеницами шелкопряда при завивке кокона, а также пауками при плетении паутины, содержит только -структуры, соединенные одиночными цепями (рис. 11). В отличие от кератина, у фиброина нет поперечных дисульфидных мостиков, он обладает очень прочен на разрыв (прочность в расчете на единицу поперечного сечения у некоторых образцов паутины выше, чем у стальных тросов). Из-за отсутствия поперечных сшивок фиброин неупруг (известно, что шерстяные ткани почти несминаемы, а шелковые легко мнутся).
Случайные конформации молекулы эластина

Строение и функция эластина
В отличие от коллагена, образующего прочные фибриллы, способные выдержать большие нагрузки, эластин (также белок межклеточного матрикса) обладает резиноподобными свойствами. Нити эластина, содержащиеся в тканях лёгких, в стенках сосудов, в эластичных связках, могут быть растянуты в несколько раз по сравнению с их обычной длиной, но после снятия нагрузки они возвращаются к свёрнутой конформации.
Эластин содержит в составе около 800 аминокислотных остатков, среди которых преобладают аминокислоты с неполярными радикалами, такие как глицин, валин, аланин. Эластин содержит довольно много пролина и лизина, но лишь немного гидроксипролина; полностью отсутствует гидроксилизин.
Наличие большого количества гидрофобных радикалов препятствует созданию стабильной глобулы, в результате полипептидные цепи эластина не формируют регулярные вторичную и третичную структуры, а принимают в межклеточном матриксе разные конформации с примерно равной свободной энергией (рис.). Это как раз тот случай строения первичной структуры, когда отсутствие одной стабильной упорядоченной конформации приводит к возникновению необходимых белку свойств.
Классификация белков по химическому строению
1 - Простые белки (протеины): альбумины, глобулины, протамины, гистоны, проламины, глютеины, склеропротеин.
Они содержат в своём составе только полипептидные цепи, состоящие из аминокислотных остатков. Их называют "простые белки". Примером простых белков могут служить основные белки хроматина - гистоны; в их составе содержится много аминокислотных остатков лизина и аргинина, радикалы которых имеют положительный заряд. Рассмотренный выше белок межклеточного матрикса эластин также относят к простым белкам.
Простые белки в свою очередь делятся на основе некоторых критериев (растворимость, состав) на ряд подгрупп: альбумины, глобулины, протамины, гистоны, проламины, глютеины, склеропротеины.
Химия простых белков
Протамины и гистоны
Данная группа белков отличается рядом характерных физико-химических свойств, своеобразием аминокислотного состава и представлена в основном балками с небольшой молекулярной массой.
Протамины обладают выраженными основными свойствами, обусловленными наличием в их составе от 60 до 85% аргинина. Так, сальмин, выделенный из молок семги, состоит на 85% из аргинина. Высоким содержанием аргинина отличается другой хорошо изученный белок – клупеин, выделенный из молок сельди: из 30 аминокислот в нем на долю аргинина приходится 21 остаток. Расшифрована первичная структура крупеина.
Протамины хорошо растворимы в воде, изоэлектрическая точка их водных растворов находится в щелочной среде. По современным представлениям, протамины, скорее всего, являются пептидами, а не белками, поскольку их молекулярная масса не превышает 5000. Они составляют белковый компонент в структуре ряда сложных белков.
Гистоны также являются белками основного характера. В их состав входят лизин и аргинин, содержание которых, не превышает 20-30%. Молекулярная масса гистонов намного больше нижнего предела молекулярной массы белков. Эти белки сосредоточены в основном в ядрах клеток в составе дезоксирибонуклеопротеинов и играют важную роль в регуляции экспрессии генов.
Проламины и глютеины
Это белки растительного происхождения, отличаются своеобразием аминокислотного состава и физико-химических свойств. Они содержатся в основном в семенах злаков (пшеница, рожь, ячмень и т.д.), составляя основную массу клейковины. Характерной особенностью проламинов является растворимость в 60-80% водном растворе этанола, в то время как все остальные простые белки в этих условиях обычно выпадают в осадок.
Наиболее изучены оризенин (белок из риса), глютеин и глиадин (из пшеницы), зеин (из кукурузы), гордеин (из ячменя).
Установлено, что проламины содержат 20-25% глютаминовой кислоты и 10-15% пролина.
Альбумины и глобулины
Эти белки относятся к белкам, широко распространенным в органвх и тканях животных. Наиболее богатыми ими белки сыворотки крови, молока, яичный белок, мышцы и др. В плазме крови человека в норме содержится около 7% белков, представленных преимущественно альбуминами и глобулинами.
Альбумины и глобулины – это глобулярные белки, различающиеся по растворимости.
Необходимо отметить, что само определение «альбумины» и «глобулины» основано на их растворимости в дистиллированной воде и полунасыщенном растворе (NH4)2SO4. Однако глобулины растворимы только в разбавленных солевых растворах.
Склеропротеины
Это фибриллярные белки, составляющие основу соединительной ткани животных и человека (коллаген, кератин, эластин и др.).
2 - Сложные белки (протеиды): липопротеиды, гликопротеиды нуклеопротеиды, фосфопротеиды, металлопротеиды, хромопротеиды.
Данные белки (протеиды) – содержат в своем составе небелковый компонент – простетическую группу.
По небелковому компоненту протеиды классифицируются на:
1) Липопротеиды – белки, содержащие липиды (хилимикроны, ЛВП, ЛНП, ЛОНП) – основная функция которых – транспорт липидов в организме.
2) Гликопротеиды – белки связанные с углеводами. Как правило эти белки являются мембранными белками, ответственными за иммунитет, группы крови,молекулярное распознавание, свертывание крови.
3) Нуклеопротеиды – белки связанные с нуклеиновыми кислотами. Например, в хромосомах ДНК намотана вокруг белков гистонов, вирусы также можно считать нуклеопротеинами.
4) Фосфопротеиды – белки, содержащие остатки фосфорной кислоты.
Примером служат: казеиноген молока, вителлин желтков яиц, ихтулин (белок икры рыб).
5) Металлопротеиды - это белки, связанные с ионами металловю
Например, белок ферритин, необходим для хранения железа. Многие металлопротеины являются ферментами, например, алкогольдегидрогеназа, окисляющая спирт, содержит ион цинка.
6) Хромопротеиды – белки, связанные с окрашенными соединениями (хромос – греч. – цвет). К таким белкам относятся – гемоглобин – белок, переносящий кислород в крови, миоглобин- белок, запасающий кислород в мышцах.
Классификация протеидов является условной.
Например, гемоглобин можно отнести и к хромопротеинам, и к металлопротеинам, а казеиноген молока – к фосфопротеинам и металлопротеинам (т.к. содержит ионы кальция).
Однако очень многие белки, кроме полипептидных цепей, содержат в своём составе небелковую часть, присоединённую к белку слабыми или ковалентными связями. Небелковая часть может быть представлена ионами металлов, какими-либо органическими молекулами с низкой или высокой молекулярной массой. Такие белки называют "сложные белки". Прочно связанная с белком небелковая часть носит название простетической группы.
Простетическая группа может быть представлена веществами разной природы. Например, белки, соединённые с гемом, носят название гемопротеины. В состав гемопротеинов, кроме уже рассмотренных выше белков гемоглобинов и миоглобина, входят ферменты - цитохромы, каталаза и пероксидаза. Гем, присоединённый к разным белковым структурам, выполняет в них характерные для каждого из белков функции (например, в составе гемоглобина переносит О2, а в составе цитохромов - электроны).
Белки, соединённые с остатком фосфорной кислоты, называют фосфопротеинами. Фосфорные остатки присоединяются сложноэфирной связью к гидроксильным группам серина, треонина или тирозина при участии ферментов, называемых протеинкиназами.
В состав белков часто входят углеводные остатки, придающие белкам дополнительную специфичность и часто уменьшающие скорость их ферментативного протеолиза. Такие белки носят название гликопротеинов. Многие белки крови, а также рецепторные белки клеточной поверхности относят к гликопротеинам.
Белки, функционирующие в комплексе с липидами, называют липопротеинами, а в комплексе с металлами - металлопротеинами.
Сложный белок, состоящий из белковой части (апопротеин) и небелковой части (простетическая группа), называют "холопротеин".
На рисунке представлена трехмерная схема жидкостно-мозаичной модели мембраны Сингера - Николсона на которой изображены глобулярные интегральные белки, погруженные в липидный бислой. Часть белков является ионными каналами, другие (гликопротеины) содержат олигосахаридные боковые цепи, участвующие в узнавании клетками друг друга и в межклеточной. Молекулы холестерола вплотную примыкают к фосфолипидным головкам и фиксируют прилегающие участки "хвостов". Внутренние участки хвостов молекулы фосфолипидов не ограничены в своем движении и ответственны за текучесть мембраны (Bretscher, 1985).
Мембрана имеет толщину 8-12 нм и состоит из бимолекулярного слоя липидов, причем гидрофобные концы молекул фосфолипидов и триглицеридов направлены внутрь, а наружу - гидрофильные головки. В двойной слой липидов встроены белки, которые пронизывают липидный слой насквозь, либо погружены в него частично. Существуют периферийные белки, покрывающие некоторые мембраны с одной или двух сторон сетью вытянутых молекул.
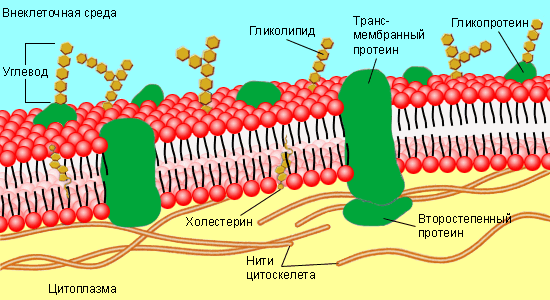
Рисунок - 1 — гликолипид, 2 — ионный канал, 3 — фосфолипид, 4 — интегральный белок, 5 — олигосахаридная боковая цепь, б — гидрофобный участок —спирали, 7 — —спиральная белковая молекула, 8 — холестерин, 9 — наружная поверхность, 10 — липидная сердцевина, 11 — внутренняя поверхность.
При этом молекулы фосфолипидов и белков находятся в непрерывном движении и взаимодействии. Липидный слой определяет основные структурные особенности биологических мембран, а белки ответственны за большинство функций мембран (транспорт, передача сигналов и т.д.). В активном состоянии мембрана имеет жидкую консистенцию, которая зависит от соотношения насыщенных и ненасыщенных жирных кислот.
В настоящее время выявлено четыре основных механизма транспорта через цитоплазматическую мембрану как клетки, так и клеточных органелл: диффузия, осмос, активный транспорт, экзо- и эндоцитоз.
Мембранные белки локализованы на поверхности мембраны или могут быть внедрены на различную глубину в гидрофобную зону. Некоторые белки пронизывают мембрану насквозь, и различные гидрофильные группы одного и того же белка обнаруживаются по обе стороны клеточной мембраны. Белки, обнаруженные в плазматической мембране, играют очень важную роль: они участвуют в образовании ионных каналов, играют роль мембранных насосов и переносчиков различных веществ, а также могут выполнять рецепторную функцию.
Лекция 5
КЛАССИФИКАЦИЯ БЕЛКОВ ПО ФУНКЦИЯМ

Белки выполняют в клетках множество биологических функций. По признаку сходства выполняемых белками функций их можно разделить на следующие большие группы.
Белки в клетках выполняют самые разнообразные функции: 1) структурная, 2) ферментативная (каталическая), 3) транспортная, 4) защитная, 5) сократительная (двигательная) функция, 6) регуляторная (гармоны), 7) рецепторная, 8) энергетическая, 9)запасающая.
Белки выполняют в клетках множество биологических функций. По признаку сходства выполняемых белками функций их можно разделить на следующие большие группы.
1 Структурные белки
Структурные белки выполняют защитную функцию (кожные покровы) или опорную – скрепляют организм в единое целое и придают ему прочность (хрящи и сухожилия)
Некоторые белки, расположенные определённым образом в тканях, придают им форму, создают опору, определяют механические свойства данной ткани.
Другие структурный беки (эластин) благодаря своему уникальному строению обеспечивает определённым тканям свойство растягиваться во всех направлениях (сосуды, лёгкие).
Белки входят в состав:
- клеточных мембран и матрикса органелл клетки;
- стенок кровеносных сосудов, хрящей, сухожилий, суставных связок – белок – эластин, легко растягивающийся в двух измерениях;
- роговые образования - волосы, ногти, рогова, перья, когти у высших животных – эти белки состоят преимущественно из белков – керотина – его основное отличие высокое содержание цистеина, образующего дисульфидные мостики, что придает высокую упругость (способность восстанавливать исходную форму после деформации) волосам, а также шерстяным тканям;
- костей – белок – оссеин;
- шелк, паутина – белок – фиброин;
- белок, участвующий в образовании тромбов – фибрин.
Для необратимого изменения формы кератинового объекта нужно вначале разрушить дисульфидные мостики с помощью восстановителя, придать новую форму, а затем вновь создать дисульфидные мостики с помощью окислителя, так делается химическая завивка волос.
При увеличении содержания остатков цистеина в кератине и, соответственно, возрастании количества дисульфидных мостиков способность к деформации исчезает, но при этом появляется высокая прочность (в рогах копытных животных и панцирях черепах содержится до 18% цистеиновых фрагментов). В организме млекопитающих содержится до 30 различных типов кератина.
Родственный кератину фибриллярный белок фиброин, выделяемый гусеницами шелкопряда при завивке кокона, а также пауками при плетении паутины, содержит только -структуры, соединенные одиночными цепями.
В отличие от кератина, у фиброина нет поперечных дисульфидных мостиков, он обладает очень прочен на разрыв (прочность в расчете на единицу поперечного сечения у некоторых образцов паутины выше, чем у стальных тросов). Из-за отсутствия поперечных сшивок фиброин неупруг (известно, что шерстяные ткани почти несминаемы, а шелковые легко мнутся).
Белок фиброин, выделяемый гусеницами шелкопряда при завивке кокона, а также пауками при плетении паутины, содержит только -структуры, соединенные одиночными цепями.
Он содержат большое количество остатков глицина, аланина и серина (каждый второй аминокислотный остаток – глицин); остатки цистеина, содержащего сульфгидридные группы, отсутствуют. Фиброин – основной компонент натурального шелка и паутины, содержит -структуры, соединенные одиночными цепями.
В состав многих белков помимо пептидных цепей входят и неаминокислотные фрагменты, по этому критерию белки делят на две большие группы — простые и сложные белки (протеиды).
Простые белки содержат только аминокислотные цепи,
сложные белки содержат также неаминокислотные фрагменты.
Эти фрагменты небелковой природы в составе сложных белков называются «простетическими группами». В зависимости от химической природы простетических групп среди сложных белков выделяют следующие классы: гликопротеиды - содержащие в качестве простетической группы ковалентно связанные углеводные остатки (в образовании связи с углеводными остатками обычно участвуют гидроксильные группы серина или треонина. Большая часть внеклеточных белков, в частности, иммуноглобулины — гликопротеиды), липопротеиды, металлопротеиды, нуклеопротеиды, фосфопротеиды (ковалентно связанные остатки фосфорной кислоты с аминокислотой (серин и треонин - в казеине молока), хромопротеиды (гемоглобин).
Трехмерная схема жидкостно-мозаичной модели мембраны Сингера- Николсона; изображены глобулярные интегральные белки, погруженные в липидный бислой. Часть белков является ионными каналами, другие (гликопротеины) содержат олигосахаридные боковые цепи, участвующие в узнавании клетками друг друга и в межклеточной. Молекулы холестерола вплотную примыкают к фосфолипидным головкам и фиксируют прилегающие участки "хвостов". Внутренние участки хвостов молекулы фосфолипидов не ограничены в своем движении и ответственны за текучесть мембраны (Bretscher, 1985).
1 — гликолипид, 2 — ионный канал, 3 — фосфолипид, 4 — интегральный белок, 5 — олигосахаридная боковая цепь, б — гидрофобный участок —спирали, 7 — —спиральная белковая молекула, 8 — холестерин, 9 — наружная поверхность, 10 — липидная сердцевина, 11 — внутренняя поверхность.
Мембрана имеет толщину 8-12 нм и состоит из бимолекулярного слоя липидов, причем гидрофобные концы молекул фосфолипидов и триглицеридов направлены внутрь, а наружу - гидрофильные головки. В двойной слой липидов встроены белки, которые пронизывают липидный слой насквозь, либо погружены в него частично. Существуют периферийные белки, покрывающие некоторые мембраны с одной или двух сторон сетью вытянутых молекул. При этом молекулы фосфолипидов и белков находятся в непрерывном движении и взаимодействии. Липидный слой определяет основные структурные особенности биологических мембран, а белки ответственны за большинство функций мембран (транспорт, передача сигналов и т.д.). В активном состоянии мембрана имеет жидкую консистенцию, которая зависит от соотношения насыщенных и ненасыщенных жирных кислот.
В настоящее время выявлено четыре основных механизма транспорта через цитоплазматическую мембрану как клетки, так и клеточных органелл: диффузия, осмос, активный транспорт, экзо- и эндоцитоз.
Мембранные белки локализованы на поверхности мембраны или могут быть внедрены на различную глубину в гидрофобную зону. Некоторые белки пронизывают мембрану насквозь, и различные гидрофильные группы одного и того же белка обнаруживаются по обе стороны клеточной мембраны. Белки, обнаруженные в плазматической мембране, играют очень важную роль: они участвуют в образовании ионных каналов, играют роль мембранных насосов и переносчиков различных веществ, а также могут выполнять рецепторную функцию.
2 Каталитическая (ферментативная) функция
Белки-ферменты катализируют протекание всех химических реакций в организме. Под действием ферментов составные компоненты пищи: белки, жиры и углеводы – расщепляются до более простых соединений, из которых затем синтезируются новые макромолекулы, необходимые организму определенного типа.
Ферменты - специализированные белки, другое их название – энзимы (en zumh греч. – в дрожжах) – это белки, обладающие каталитической активностью, они способны увеличивать скорости биохимических реакций в тысячи раз.
Они обеспечивают фиксацию углерода при фотосинтезе, разложение перекиси водорода может катализироваться как просто ионами железа, так и железосодержащим белком каталазой.
Ферменты принимают участие и во многих биохимических процессах синтеза, например, в синтезе белков (одни белки помогают синтезировать другие).
Ферменты не только высокоэффективные, но и селективные катализаторы, (направляют реакцию строго в заданном направлении). В их присутствии реакция проходит практически со 100%-ным выходом без образования побочных продуктов и при этом условия протекания – мягкие: обычное атмосферное давление и температура живого организма.
Для сравнения, синтез аммиака из водорода и азота в присутствии катализатора – активированного железа – проводят при 400–500°С и давлении 30 МПа, выход аммиака 15–25% за один цикл.
В настоящее время известно около 2000 различных ферментов, ускоряющих различные химические реакции.
Например, протеолитический фермент трипсин разрушает в белках пептидные связи, образованные карбоксильной группой основных аминокислот - аргинина или лизина. Фермент рибонуклеаза расщепляет фосфоэфирную связь между нуклеотидами в полинуклеотидной цепи.
Благодаря набору ферментов в клетках превращения поступающих в них веществ протекают не хаотично, а в строго определённых направлениях.
Интенсивное исследование ферментов началось в середине 19 в., сейчас изучено более 2000 различных ферментов, это самый многообразный класс белков.
Названия ферментов составляют следующим образом: к наименованию реагента, с которым взаимодействует фермент, или к названию катализируемой реакции добавляют окончание -аза,
Например, аргиназа разлагает аргинин, декарбоксилаза катализирует декарбоксилирование, т.е. отщепление СО2 от карбоксильной группы:
– СООН – СН + СО2
Часто, для более точного обозначения роли фермента в его названии указывают и объект, и тип реакции, например, алкогольдегидрогеназа – фермент, осуществляющий дегидрирование спиртов.
Для некоторых ферментов, открытых достаточно давно, сохранилось историческое название (без окончания –аза), например, пепсин (pepsis, греч. пищеварение) и трипсин (thrypsis греч. разжижение), эти ферменты расщепляют белки.
Для систематизации ферменты объединяют в крупные классы, в основу классификации положен тип реакции, классы именуют по общему принципу – название реакции и окончание – аза.
Далее перечислены некоторые из таких классов.
Оксидоредуктазы – ферменты, катализирующие окислительно-восстановительные реакции. Входящие в этот класс дегидрогеназы осуществляют перенос протона, например алкогольдегидрогеназа (АДГ) окисляет спирты до альдегидов, последующее окисление альдегидов до карбоновых кислот катализируют альдегиддегидрогеназы (АЛДГ). Оба процесса происходят в организме при переработке этанола в уксусную кислоту.
Наркотическим действием обладает не этанол, а промежуточный продукт ацетальдегид, чем ниже активность фермента АЛДГ, тем медленнее проходит вторая стадия – окисление ацетальдегида до уксусной кислоты и тем дольше и сильнее проявляется опьяняющее действие от приема внутрь этанола. Анализ показал, что более чем у 80% представителей желтой расы относительно низкая активность АЛДГ и потому заметно более тяжелая переносимость алкоголя. Причина такой врожденной пониженной активности АЛДГ состоит в том, что часть остатков глутаминовой кислоты в молекуле «ослабленной» АЛДГ заменена фрагментами лизина (табл. 1).
Трансферазы – ферменты, катализирующие перенос функциональных групп, например, трансиминаза катализирует перемещение аминогруппы.
Гидролазы – ферменты, катализирующие гидролиз. Упомянутые ранее трипсин и пепсин осуществляют гидролиз пептидных связей, а липазы расщепляют сложноэфирную связь в жирах:
–RС(О)ОR1 +Н2О –RС(О)ОН + НОR1
Лиазы – ферменты, катализирующие реакции, которые проходят не гидролитическим путем, в результате таких реакций происходит разрыв связей С-С, С-О, С-N и образование новых связей. Фермент декарбоксилаза относится к этому классу.
Изомеразы – ферменты, катализирующие изомеризацию, например, превращение малеиновой кислоты в фумаровую, это пример цис – транс изомеризации.
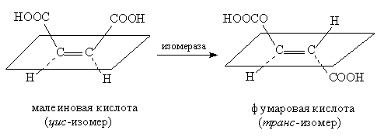
В работе ферментов соблюдается общий принцип, в соответствии с которым всегда есть структурное соответствие фермента и реагента ускоряемой реакции. По образному выражению одного из основателей учения о ферментах Э.Фишера, реагент подходит к ферменту, как ключ к замку.
В связи с этим каждый фермент катализирует определенную химическую реакцию или группу реакций одного типа.
Иногда фермент может действовать на одно единственное соединение, например, уреаза (греч. uron– моча) катализирует только гидролиз мочевины:
(H2N)2C = O + H2O = CO2 + 2NH3
Наиболее тонкую избирательность проявляют ферменты, различающие оптически активные антиподы – лево- и правовращающие изомеры. L-аргиназа действует только на левовращающий аргинин и не затрагивает правовращающий изомер. L-лактатдегидрогеназа действует только на левовращающие эфиры молочной кислоты, так называемые лактаты (lactis лат. молоко), в то время как D-лактатдегидрогеназа расщепляет исключительно D-лактаты.
Большая часть ферментов действует не на одно, а на группу родственных соединений, например, трипсин «предпочитает» расщеплять пептидные связи образованные лизином и аргинином.
Каталитические свойства некоторых ферментов, таких как гидролазы, определяются исключительно строением самой белковой молекулы, другой класс ферментов – оксидоредуктазы (например, алкогольдегидрогеназа) могут проявлять активность только в присутствии связанных с ними небелковых молекул – витаминов, активирующих ионов Mg, Са, Zn, Мn и фрагментов нуклеиновых кислот.
В работе ферментов соблюдается общий принцип, в соответствии с которым всегда есть структурное соответствие фермента и реагента ускоряемой реакции. По образному выражению одного из основателей учения о ферментах Э.Фишера, реагент подходит к ферменту, как ключ к замку.
В связи с этим каждый фермент катализирует определенную химическую реакцию или группу реакций одного типа.
Иногда фермент может действовать на одно единственное соединение, например, уреаза (uron греч. – моча) катализирует только гидролиз мочевины:
(H2N)2C = O + H2O = CO2 + 2NH3
3 Транспортные белки
Некоторые белки способны присоединять и переносить различные вещества.
Часто в комплексе с белками переносятся молекулы, плохо растворимые в воде.
Транспортные белки участвуют также в переносе гидрофильных веществ через гидрофобные мембраны. Так как транспортные белки обладают свойством специфичности взаимодействия с лигандами, их набор в клеточной мембране определяет, какие гидрофильные молекулы могут пройти в данную клетку. С помощью белков-переносчиков в клетку проникают глюкоза, аминокислоты, ионы и другие молекулы.
а) Альбумины крови - транспортируют жирные кислоты и билирубин (продукт распада тема), а гемоглобин эритроцитов участвует в переносе О2 от лёгких к тканям.
Стероидные гормоны переносятся в крови специфическими транспортными белками.
б) Глобулины - ионы металлов и гормоны, гемоглобин — белок, определяющий красный цвет крови, присоединяет кислород и переносит его во все органы и ткани и удаляет углекислый газ.
в) Очень многие вещества, включая лекарственные препараты, не могут сами путешествовать по организму или по клетке – им нужны для этого белки-переносчики.
г) Многие ионы не могут проникать через клеточную мембрану – для этого существуют специальные белки переносчики, многие из которых также участвуют в создании разности потенциалов и проведении электрического возбуждения по нервным волокнам и мышцам.
д) Молекулы белков, входящие в состав плазматической мембраны, принимают участие в транспортировке веществ в клетку.
Существуют также транспортные белки, способные связывать жиры, глюкозу, аминокислоты и переносить их как внутрь, так и вовне клеток.
Транспортные белки особого типа не переносят сами вещества, а выполняют функции «транспортного регулировщика», пропуская определенные вещества сквозь мембрану (внешнюю стенку клетки). Такие белки чаще называют мембранными. Они имеют форму пустотелого цилиндра и, встраиваясь в стенку мембраны, обеспечивают перемещение некоторых полярных молекул или ионов внутрь клетки. Пример мембранного белка – порин (следующий слайд).
БЕЛОК ПОРИН - состоящий преимущественно из b-структур, представляет собой пустотелый цилиндр. Встраиваясь в мембрану (стенку) клетки, он пропускает внутрь клетки определенные органические молекулы. На рисунке показана одна из таких молекул, аминоспирт H2NC(CH2OH)3 – вещество, участвующее в передаче нервных импульсов.
4 Защитные белки
Защитные белки позволяют уберечь организм от вторжения атакующих его бактерий, вирусов и от проникновения чужеродных белков (обобщенное название чужеродных тел – антигены).
Роль защитных белков выполняют иммуноглобулины (другое их название – антитела), они распознают антигены, проникшие в организм, и прочно связываются с ними.
В организме млекопитающих, включая человека, есть пять классов иммуноглобулинов: M, G, A, D и E, их структура, как следует из названия, глобулярная, кроме того, все они построены сходным образом. Молекулярная организация антител показана на слайде на примере иммуноглобулина класса G. Молекула содержит четыре полипептидные цепи, объединенные тремя дисульфидными мостиками S-S (на слайде они показаны с утолщенными валентными связями и крупными символами S), кроме того, каждая полимерная цепь содержит внутрицепные дисульфидные перемычки.
Две большие полимерные цепи (выделены синим цветом) содержат 400–600 аминокислотных остатков.
Две другие цепи (выделены зеленым цветом) почти вдвое короче, они содержат приблизительно 220 аминокислотных остатков. Все четыре цепи расположены таким образом, что концевые H2N-группы направлены в одну сторону.
После контакта организма с чужеродным белком (антигеном), клетки иммунной системы начинают вырабатывать иммуноглобулины (антитела), которые накапливаются в сыворотке крови. На первом этапе основную работу совершают участки цепей, содержащие концевые H2N (на рис. 27 соответствующие участки отмечены светло-синим и светло-зеленым цветом). Это области захвата антигенов. В процессе синтеза иммуноглобулина эти участки формируется таким образом, чтобы их строение и конфигурация максимально соответствовали структуре приблизившегося антигена (как ключ к замку, подобно ферментам, но задачи в данном случае иные). Таким образом, для каждого антигена в качестве иммунного ответа создается строго индивидуальное антитело. Столь «пластично» изменять строение в зависимости от внешних факторов, помимо иммуноглобулинов, не может ни один известный белок. Ферменты решают задачу структурного соответствия реагенту иным путем – с помощью гигантского набора разнообразных ферментов в расчете на все возможные случаи, а иммуноглобулины каждый раз заново перестраивают «рабочий инструмент». Сверх того, шарнирный участок иммуноглобулина обеспечивает двум областям захвата некоторую независимую подвижность, в результате молекула иммуноглобулина может «найти» сразу два наиболее удобных для захвата участка в антигене с тем, чтобы его надежно зафиксировать, это напоминает действия ракообразного существа.
Далее включается цепь последовательных реакций иммунной системы организма, подключаются иммуноглобулины других классов, в результате происходит дезактивация чужеродного белка, а затем уничтожение и удаление антигена (постороннего микроорганизма или токсина).
После контакта с антигеном максимальная концентрация иммуноглобулина достигается (в зависимости от природы антигена и индивидуальных особенностей самого организма) в течение нескольких часов (иногда нескольких дней). Организм сохраняет память о таком контакте, и при повторной атаке таким же антигеном иммуноглобулины накапливаются в сыворотке крови значительно быстрее и в большем количестве – возникает приобретенный иммунитет.
Приведенная классификация белков носит в определенной степени условный характер, например белок тромбин, упомянутый среди защитных белков, по существу представляет собой фермент, катализирующий гидролиз пептидных связей, то есть, относится к классу протеаз.
К защитным белкам часто относят белки змеиного яда и токсичные белки некоторых растений, поскольку их задача – уберечь организм от повреждений.
Есть белки, функции которых настолько уникальны, что это затрудняет их классификацию. Например, белок монеллин, содержащийся в одном из африканских растений, – очень сладкий на вкус, он стал предметом изучения как нетоксичное вещество, которое может быть использовано вместо сахара для предотвращения ожирения. Плазма крови некоторых антарктических рыб содержит белки со свойствами антифриза, который предохраняет кровь этих рыб от замерзания.
Защитными свойствами обладают белки свёртывающей системы крови, например фибриноген, тромбин. Они участвуют в формировании тромба, который закупоривает повреждённый сосуд и препятствует потере крови.
5 Сократительные и двигательные белки придают организму способность сокращаться, изменять форму и перемещаться, прежде всего, речь идет о мышцах. 40% от массы всех белков, содержащихся в мышцах, составляет миозин (mys, myos, греч. – мышца). Его молекула содержит одновременно фибриллярную и глобулярную часть.
Такие молекулы объединяются в крупные агрегаты, содержащие 300–400 молекул.
При изменении концентрации ионов кальция в пространстве, окружающем мышечные волокна, происходит обратимое изменение конформации молекул – изменение формы цепи за счет поворота отдельных фрагментов вокруг валентных связей. Это приводит к сокращению и расслаблению мышц, сигнал для изменения концентрации ионов кальция поступает от нервных окончаний в мышечных волокнах. Искусственное сокращение мышц можно вызвать действием электрических импульсов, приводящих к резкому изменению концентрации ионов кальция, на этом основана стимуляция сердечной мышцы для восстановления работы сердца.
Благодаря скольжению относительно друг друга актиновых (актины) и миозиновых (миозины) протофибрилл происходит сокращение мышц, а также немышечные внутриклеточные сокращения. Движение ресничек и жгутиков связано со скольжением относительно друг друга микротрубочек, имеющих белковую природу.
Некоторые арктические и антарктические рыбы содержат в крови белки – антифризы, предотвращающие ее замораживание.
Некоторые белки при выполнении своих функций наделяют клетку способностью либо сокращаться, либо передвигаться. К таким белкам относят актин и миозин - фибриллярные белки, участвующие в сокращении скелетных мышц. Другой пример таких белков - тубулин, из которого построены клеточные органеллы - микротрубочки. Микротрубочки в период деления клетки регулируют расхождение хроматид. Микротрубочки - важные элементы ресничек и жгутиков, с помощью которых клетки передвигаются.
Однако существует большое количество белков, имеющих уникальные функции, которые не вошли в эту довольно простую классификацию.
6 Регуляторные белки, чаще называемые гормонами, участвуют в различных физиологических процессах.
К регуляторным белкам относят большую группу белковых гормонов, участвующих в поддержании постоянства внутренней среды организма, которые воздействуют на специфические клетки-мишени.
Многие гормоны являются олигопептидами или белками (например, инсулин, глюкагон [антагонист инсулина], адренокортикотропный гормон и др.).
Гормон инсулин состоит из двух -цепей, соединенных дисульфидными мостиками.
Инсулин – гормон, образующийся в клетках островков Лангерганса в поджелудочной железе. Он играет важнейшую роль в метаболизме глюкозы в крови.
Кроме того, к регуляторным относят белки, присоединение которых к другим белкам или иным структурам клетки регулирует их функцию. Например, белок кальмодулин в комплексе с четырьмя ионами Са2+ может присоединяться к некоторым ферментам, меняя их активность.
Регуляторные ДНК-связывающие белки, присоединяясь в определённые моменты к специфичным участкам ДНК, могут регулировать скорость считывания генетической информации.
В гипофизе мозга синтезируется гормон, регулирующий рост организма. Существуют регуляторные белки, контролирующие биосинтез различных ферментов в организме.
На рисунке показан - БЕЛОК ИНСУЛИН - в виде объемной модели и в форме третичной структуры. Состоит из двух -спиральных цепей, связанных двумя дисульфидными мостиками (сравни с рис. 2, где его строение показано схематически)
МОЛЕКУЛА ИНСУЛИНА, построенная из 51 аминокислотного остатка, фрагменты одинаковых аминокислот отмечены соответствующей окраской фона. Содержащиеся в цепи остатки аминокислоты цистеина (сокращенное обозначение ЦИС) образуют дисульфидные мостики –S-S-, которые связывают две полимерных молекулы, либо образуют перемычки внутри одной цепи.
7 Рецепторная (сигнальная) функция белков
Сигнальные молекулы (гормоны, нейромедиаторы) действуют на внутриклеточные процессы через взаимодействие со специфическими белками-рецепторами. Так, гормоны, циркулирующие в крови, находят клетки-мишени и воздействуют на них, специфично связываясь с белками-рецепторами, обычно встроенными в клеточную мембрану. Для гидрофобных регуляторных молекул, проходящих через клеточную мембрану, рецепторы локализуются в цитоплазме клеток.
Некоторые белки, встроенные в клеточную мембрану, способны изменять свою структуру под воздействием внешней среды.
Так происходит прием сигналов извне и передача информации в клетку.
Примером может служить фитохром — светочувствительный белок, регулирующий фотопериодическую реакцию растений, и опсин — составная часть родопсина пигмента - , интегральный мембранный белок, находящегося в клетках сетчатки глаза.
Фитохром (от Фито... и греч. chroma – цвет, краска) голубой пигмент из группы сложных белков – хромопротеидов; присутствует в клетках фотосинтезирующих организмов. Впервые обнаружен американсканским биохимиком У. Батлером в 1959 в семядолях проростков турнепса, выращенных в темноте.
Голубоватые фитохромы относятся к фотосинтетически неактивным пигментам.
Однако установлено, что под контролем фитохрома находятся синтезы биополимеров (ДНК, РНК, белков), системы биосинтеза хлорофилла, каротиноидов, антоцианов, органических фосфатов, витаминов. Ф. ускоряет катаболитический распад полисахаридов, жиров и резервных белков, активирует клеточное дыхание и Окислительное фосфорилирование.
Ферменты существует в двух взаимопревращаемых формах – Ф660 и Ф730, различных по спектрам поглощения. Под действием красного света с длина волны = 660 нм неактивный Ф660 превращается в активный Ф730. Обратное превращение происходит либо в темноте, либо при освещении красным светом с = 730 нм. Считают, что эти взаимопревращения обусловлены цис-транс-изомеризацией хромофора Ф. и конформационными перестройками белка.
Сигнальные молекулы (гормоны, нейромедиаторы) действуют на внутриклеточные процесс через взаимодействие со специфическими белками рецепторами.
Гормоны, циркулирующие в крови, находят клетки-мишени и воздействуют на них, специфично связываясь с белками-рецепторами, обычно встроенными в клеточную мембрану. Для гидрофобных регуляторных молекул, проходящих через клеточную мембрану, рецепторы локализуются в цитолазме клеток.
Сигнальные молекулы (гормоны, нейромедиаторы) действуют на внутриклеточные процессы через взаимодействие со специфическими белками-рецепторами. Так, гормоны, циркулирующие в крови, находят клетки-мишени и воздействуют на них, специфично связываясь с белками-рецепторами, обычно встроенными в клеточную мембрану. Для гидрофобных регуляторных молекул, проходящих через клеточную мембрану, рецепторы локализуются в цитоплазме клеток.
Наиболее важными из них являются фитохромы А и В (phyA and phyB). Фитохром А
выполняет множество различных фоторегуляторных функций. При его участии происходит стимулирование и ингибирование проростания семян, индукция де-этиоляции, регуляция синтеза различных ферментов, регуляция развития корня, стимуляция цветения и регуляция циркадных ритмов.
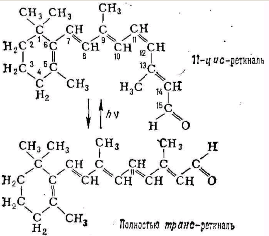
Цикл основных изменений родопсина в палочках сетчатки
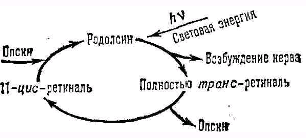
РОДОПСИН (от греч. rhodon - роза и opsis - зрение), зрительный пурпур, осн. зрительный пигмент палочек сетчатки позвоночных (кроме нек-рых рыб и земноводных на ранних стадиях развития) и беспозвоночных животных.
По химич. природе родопсин - сложный белок (хромопротеид), в состав которого входит 11-цис-ретиналь (хромофорная группа), гликопротеид, т. е. белок, соединённый с сахарами, и липиды (т. н. опсино-вая часть). Мол. масса родопсина позвоночных ок. 40 000, головоногих моллюсков-ок. 70 000. Р.- осн. структурно-функциональный компонент наружного сегмента палочек (см. Зрение, Сетчатка, Фоторецепторы).
Зрительный акт начинается поглощением Р. кванта света (максимум спектра поглощения Р.- ок. 500 нм). При этом происходит изомеризация 11-цис-ретиналя в полностью транс-форму (см. формулы), что приводит к постепенному разложению (фотолизу) молекулы Р., изменению ионного транспорта в фоторецепторе и возникновению электрич. сигнала, к-рый передаётся нервным элементам сетчатки. Регенерация Р. осуществляется или путём синтеза из 11-цис-ретиналя и освободившегося после фотолиза опсина, или при поглощении второго кванта одним из промежуточных продуктов фотолиза, а также в процессе синтеза новых дисков наружного сегмента сетчатки (последний путь для палочек основной).
В клеточных оболочках некоторыхрых галофильных бактерий обнаружен пигмент, в состав которого также входят ретиналь, гликопротеид и липиды. Этот бактериальный радапсин (структура его окончательно не установлена), по-видимому, участвует в фотосинтезе наряду с др. пигментами бактерий.
Особое значение для действия фитохрома имеет его обратимость: этот хромопротеид (сложный белок, содержащий, кроме аминокислот, также окрашивающие компоненты) встречается в двух формах, способных преобразовываться одна в другую.
Голубой фитохром 660 (Ф 660) имеет максимум поглощения в светло-красной области спектра с длиной волны 660 нм, а зелено-голубой фитохром 730 (Ф 730) - в темно-красной области спектра с длиной волны 730 нм. 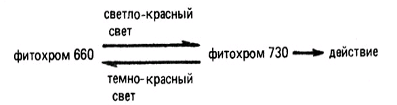
При освещении светло-красным светом неактивный Ф 660 превращается в физиологически активный Ф 730, а при освещении темно-красным светом происходит превращение Ф 730 в Ф 660.
8 Пищевые и запасные белки, как следует из названия, служат источниками внутреннего питания, чаще для зародышей растений и животных, а также на ранних стадиях развития молодых организмов.
К пищевым белкам относят альбумин – основной компонент яичного белка, а также казеин – главный белок молока.
Под действием фермента пепсина казеин в желудке створаживается, это обеспечивает его задержку в пищеварительном тракте и эффективное усвоение. Казеин содержит фрагменты всех аминокислот, необходимых организму.
В ферритине, который содержится в тканях животных, запасены ионы железа.
К запасным белкам относят также миоглобин, по составу и строению напоминающий гемоглобин. Миоглобин сосредоточен, главным образом, в мышцах, его основная роль – хранение кислорода, который ему отдает гемоглобин. Он быстро насыщается кислородом (намного быстрее, чем гемоглобин), а затем постепенно передает его различным тканям при последующей физической нагрузке и кислородной недостаточности его высвободить..
Все это разнообразие функций проистекает из очень простого набора 20 аминокислот, из которых построена полипептидная цепь белка. Именно разное количество и разные сочетания этих аминокислот в цепи и определяет уникальность того или иного белка.
9 Энергетическая функция
Белки могут служить источником энергии в клетке (после их гидролиза).
Энергетическая функция
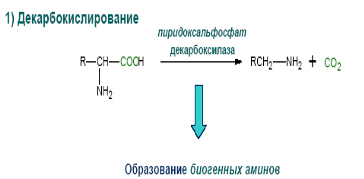
Обычно белки расходуются на энергетические нужды в крайних случаях, когда исчерпаны запасы углеводов и жиров.
При отщеплении от аминокислот карбоксильной группы образуются амины:
Белки, после гидролиза первичной структуры (до аминокислот) могут подвергаться дезаминированию.
Например, при окислении аминокислоты происходит отщепление аммиака с образованием кетонокислоты, которая распадается на альдегид и углекислоту.
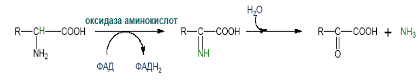
Кетокислота Альдегид
Процесс дезаминирования и декарбоксилирования идут в мягких условиях под влиянием оксидазами аминокислот.
Аминокислоты реагируют также с кетокислотами и вступают в реакцию трансаминирования.

Трансаминирование — реакция переноса -аминогруппы с АК на -кетокислоту, в результате чего образуются новая -кетокислота и новая АК. Процесс трансаминирования легко обратим, при нем общее количество АК в клетке не меняется.
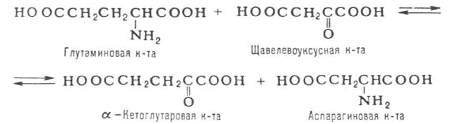
На первой стадии реакции аминогруппа реагирует с карбонильной группой кетокислоты, образую нестойкое иминопроизводное.
Переаминирование, катализируемое ферментами трансаминазами (аминотрансферазами), представляет собой взаимопревращение пары аминокислот и пары кетокислот.
Реакции трансаминирования (переаминирование) обеспечивают синтез и распад амино- и кетокислот, перераспределение аминного азота в тканях организма.
У человека найдено более 10 аминотрансфераз, которые локализуются в цитоплазме и митохондриях клеток. В реакции трансаминирования вступают почти все АК, за исключением лизина, треонина и пролина.
Наибольшая роль переаминирования играет в биохимии в процессах метаболизма азотистых оснований в тканях животных и растений. Заключается в переносе аминогруппы от молекулы a-аминокислоты в молекулу a-кетокислоты, как правило с участием ферментов - аминотрансфераз (трансаминаз), например по реакции: (превращение глутаминовой кислоты в аспарагиновую)
В живых организмах на реакциях такого типа основываются синтез и диссимиляция аминокислот.
Аминотрансферазы (более 50 разновидностей) содержат в качестве кофсрмента производные витамина В6-пири-доксаль-5'-фосфат (ф-ла I) и пиридоксамин-5'-фосфат (II). В основе каталитич. активности пиридоксаль-5'-фосфата лежит способность его формильной группы образовывать с аминокислотами шиффовы основания, легко гидролизующиеся до пиридоксамин-5'-фосфата и a-кетокислоты. Общая схема П. с участием этих ферментов представляет собой сумму двух полуреакций:
Вначале, АК передает свою аминогруппу на пиродоксальфосфат. АК при этом превращается в кетокислоту, а пиродоксальфосфат - в пиридоксаминфосфат.
Затем, реакции идут в обратную сторону: но уже другая кетокислота, принимает аминогруппу от пиридоксаминфосфата и превращается в новую АК, а пиридоксаминфосфат в пиродоксальфосфат.
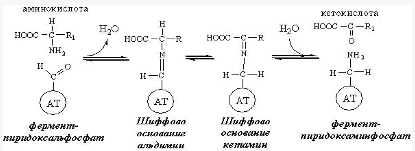
Механизм переаминирования
КЛАССИФИКАЦИЯ БЕЛКОВ
По стороению:
1) Простые (протеины) – состоят только из остатков аминокислот;
2) Сложные (протеиды) – при гидролизе образуют аминокислоты и другие соединения:
а) нуклеопротеиды: белок+нуклеиновая кислота, растворимы в щелочах, не растворимы в кислотах;
б) фосфопротеиды: белок + остаток фосфорной кислоты. Денатурируют при действии кислот (казеин молока);
в) глюкопротеиды: белок + углерод. Нерастворимы в воде, растворимы в щелочах, нейтральны (слизь);
г) хромопротеиды: белок + красящее вещество (гемоглобин).
По растворимости:
- Склеропротеины – нерастворимы в воде;
- Альбумины – растворимы в воде;
- Глобулины – растворимы в 70% этаноле;
Гистоны и протамины – растворимы в щелочах
СВОЙСТВА БЕЛКОВ
1)Некоторые белки растворяются в воде (альбумины). Альбумин связывает и переносит билирубин, жирные кислоты, различные гормоны, ионы хлора, кальция, лекарственные вещества. Альбумины — белки, растворимые в воде и осаждающиеся в насыщенном растворе сульфата аммония; характеризуются относительно небольшим молекулярным весом (15000— 65000);
В органических растворителях (абсолютном спирте, эфире, бензоле и др.) белки нерастворимы.
Растворы белков обладают свойствами, характерными для растворов высокомолекулярных соединений. В отличие от низкомолекулярных соединений белки не проходят через полупроницаемые перегородки (например, мембраны из коллодия, целлофана и т.д.). Этим их свойством пользуются для очистки белков от низкомолекулярных органических и неорганических веществ.
Растворы белков очень нестойки. Это связано со способностью молекул белка гидратироваться.
Любой фактор снижающий гидратацию белка понижает его растворимость и способствует выделению его из раствора.
К таким водоотнимающим веществам относится спирт, ацетон, концентрированные растворы сульфата натрия, аммония, магния, хлористого натрия и т. д. Различные белки осаждаются из растворов при различных концентрациях осадителей (спирта, солей, ацетона). Этим свойством широко пользуются для разделения белков.
Как и аминокислоты белки являются амфотерными соединениями – они образуют соли с кислотами и с основаниями.
Так же как и аминокислоты, белки образуют в кислой среде катионы, а в щелочной – анионы, которые, при наложении электрического поля (тока) мигрируют к аноду или катоду (данное явление называется – электрофоре).
Все белки растворимы в щелочах, многие растворимы в кислотах и разбавленных растворах солей.
В органических растворителях (абсолютном спирте, эфире, бензоле и др.) белки нерастворимы.
При определенной величине рН раствора в молекуле белка устанавливается равенство положительных и отрицательных зарядов. В зависимости от числа и природы основных и кислых групп в белке это равенство (называемое ИЗОЭЛЕКТРИЧЕСКОЙ точкой) достигается, (у разных белков)при разных концентрациях водородных ионов (Н+). У белков содержащих больше основных аминокислот, изоэлектрическая точка лежит в щелочной зоне, а у кислых в кислой зоне.
Так например изоэлектрическая точка ( рI ) белков … лежит при следующих рН:
Желатин -4,2; Казеин – 4,6; Альбумин яйца – 4,8; Альбумин сыворотки крови -4,8; Гемоглобин – 6,8; Гистон зобной железы 8,7.
В изоэлектрической точке ионы белка не переносятся ни к аноду, ни к катоду. В этой точке достигают своего минимального значения такие свойства белков как: набухание, вязкость, электропроводность.
Резко падает растворимость белка и увеличивается его способность к свертыванию.
Боковые цепи аминокислот (радикал) могут быть гидрофобными или гидрофильными, что придает белкам соответствующие свойства, которые проявляются при образовании вторичной, третичной и четвертичной структур белка.
ГИДРОЛИЗ И ДЕНАТУРАЦИЯ БЕЛКОВ
Гидролиз белков – разрушение первичной структуры белка под действием кислот, щелочей или ферментов, приводящее к образованию -аминокислот, из которых он был составлен.
Денатурация – это процесс нарушения высших уровней организации белковой молекулы (вторичного, третичного, четвертичного) под действием различных факторов.
При этом полипептидная цепь разворачивается и находится в растворе в развернутом виде или в виде беспорядочного клубка.
При денатурации утрачивается гидратная оболочка и белок выпадает в осадок и при этом утрачивает нативные свойства.
Денатурацию вызывают физические факторы: температура, давление, механические воздействия, ультразвуковые и ионизирующие излучения; химические факторы: кислоты, щелочи, органические растворители, алкалоиды, соли тяжелых металлов.
Различают 2 вида денатурации:
- обратимая денатурация – ренатурация или ренактивация – это процесс, при котором денатурированный белок, после удаления денатурирующих веществ вновь самоорганизуется в исходную структуру с восстановлением биологической активности.
- необратимая денатурация – это процесс, при котором биологическая активность не восстанавливается после удаления денатурирующих агентов.
РЕАКЦИИ ХАРАКТЕРНЫЕ ДЛЯ БЕЛКОВ
- Органическими растворителями: спирт, ацетон;
- Концентрированными растворами минеральных солей;
- Солями тяжелых металлов: хлорной ртутью (НgCI2); (CuSO4); (CH3COOCu); (CH3COOPb) ;
- Кислотами: ТХУК, НNO3, CH3COOH;
- Коллоидными растворами гидратов окиси железа [Fe(OH)3] алюминия [Al(OH)3].
При осаждении белков спиртом, ацетоном и концентрированными растворами минеральных солей белки не теряют способность снова переходить в раствор. Это используют для выделения и разделения белков. При действии остальных осадителей белки в значительной степени изменяют свои свойства (денатурируются), например, теряют способность растворяться в воде и солевых растворах.
Белки можно денатурировать и физическими методами: нагреванием до температуры выше 60-70°C, облучением ультрафиолетовыми лучами, применением давления свыше 2500-3000 атм.
Примером необратимого осаждения (денатурации) является свертывание яичного белка при нагревании, свертывание крови.
Белки дают ряд цветных реакций.
Лекция 6
УГЛЕВОДЫ
Углеводы наряду с белками - наиболее распространенные соединения, они содержатся в клетках растительных и животных организмов, используются в процессе их жизнедеятельности, и по массе составляют основную часть органического вещества на Земле. Самым богатым источником углеводов служат растения: до 80% сухой массы тканей растений составляют углеводы.
В организмах животных и человека их значительно меньше; наиболее богаты углеводами печень (5 - 10%), скелетные мышцы (1 - 3%), сердечная мышца (-0,5%), головной мозг (0,2%).
Углеводами называют очень большое число соединений, обладающих различной химической структурой и биологическими функциями. Название происходит от слов «уголь» и «вода».
С точки зрения химии углеводы являются органическими веществами, содержащими неразветвлённую цепь из нескольких атомов углерода, карбонильную группу, а также несколько гидроксильных групп.
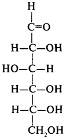
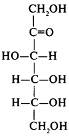
D-глюкоза D-фруктоза
Изучение углеводов показало, что существуют соединения, которые по всем свойствам нужно отнести в группу углеводов, хотя они имеют состав не точно соответствующий формуле СмH2пОп , но для которых данный термин употребляется до настоящего времени.
Тем не менее старинное название «углеводы», сохранилось до наших дней, хотя наряду с этим названием для обозначения рассматриваемой группы веществ иногда применяют и более новое название - глициды.
Усвояемые углеводы
Известно более 70 различных моносахаридов, однако только некоторые из них используются в питании. Наибольшей пищевой ценностью обладают альдозы (глюкоза, галактоза, манноза, ксилоза), а также кетозы (фруктоза). Потребление глюкозы и фруктозы – двух наиболее распространенных в природе моносахаридов – достигает 20% общего потребления углеводов. Из кишечника углеводы всасываются в кровь только в виде глюкозы и фруктозы. Глюкозу в качестве питательного материала в организме человека используют в основном нервные клетки, мозговое вещество почек и эритроциты.
Депонируется глюкоза в виде гликогена печени (100 г) и мышц (250 г). В организме постоянный уровень концентрации глюкозы в крови поддерживается с помощью гормонов поджелудочной железы – инсулина и глюкагона.
Животные организмы не способны синтезировать углеводы и получают их из растительных источников.
Углеводы образуются в растениях в процессе фотосинтеза из диоксида углерода и воды с использованием солнечной энергии. В самом общем виде фотосинтез может быть представлен в виде следующей реакции:
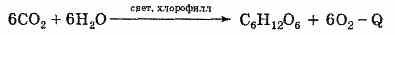
Эта энергия высвобождается в животных организмах в результате метаболизма углеводов, заключающегося с химической точки зрения в их окислении:
Сх(Н2О)у + хО2 хСО2 + уH2О + энергия
Углеводы можно рассматривать как своеобразное «депо» энергии. Часть выделяющейся при метаболизме углеводов энергии превращается в теплоту, а часть – в новую химическую форму, запасаемую в АТФ и затем расходуемую в процессах жизнедеятельности (сокращение мышечных волокон, передача нервного импульса и др.).
Суммарный энергетический эффект аэробного распада глюкозы до конечных продуктов составляет 38 моль АТР (всего образуется 40 моль АТФ, но 2 моля используется для самой реакции).
Анаэробный и аэробный гликолиз энергетически неравноценны. Образование двух моль лактата из глюкозы в ходе анаэробного гликолиза сопровождается синтезом всего двух моль АТР. Анаэробный гликолиз, несмотря на небольшой энергетический эффект, является основным источником энергии для скелетных мышц в начальном периоде интенсивной работы, то есть в условиях, когда снабжение кислородом ограничено. Кроме того, зрелые эритроциты извлекают энергию за счет анаэробного окисления глюкозы, потому что не имеют митохондрий.
КЛАССИФИКАЦИЯ УГЛЕВОДОВ
Классификация углеводов ступенчатая.
В первую очередь, отмечается количество атомов углерода углеводородной части молекулы - тетрозы, пентозы, гексозы, гептозы и т.д. Наиболее распространенными являются пентозы и гексозы. Нормальные углеводы структурно расходуют один углеродный атом на карбонильную группу, при всех остальных углеродных атомах находится по одной гидроксильной группе.
Классификация углеводов основана на их способности гидролизоваться. Углеводы разделяются на простые и сложные. Простые углеводы иначе называются моносахаридами. Сложные подразделяют на олигосахариды и полисахариды.
Итак, все углеводороды можно разделить на три больших класса: моносахариды, олигосахариды и полисахариды.
1) Моносахариды (простые сахара, иначе называют мономеры) – это структурная единица любых углеводов, они не могут быть гидролизованы до более простых форм углеводов. По химическому составу монозы являются либо полигидроксиальдегидами, либо полигидроксикетонами. Моносахариды, в состав которых входит альдегидная группа ( ), называют альдозами, а кетонная( ) - кетозами.
2) Олигосахариды – это олигомеры, состоящие из нескольких (не более 10) мономеров – моносахаридов, связанных между собой гликозидной связью. Они делятся по числу моносахаридов в молекуле на дисахариды (или биозы), трисахариды (или триозы), тетрасахариды (тетрозы), пентасахариды (или пентозы), гексасахариды (или гексаозы) и т.д.
3) Полисахариды – название класса сложных высокомолекулярных углеводов, молекулы которых состоят из десятков, сотен или тысяч мономеров – остатков моносахаридов.
Полисахариды (дисахариды) – это углеводы, которые при гидролизе дают «n» одинаковых или различные молекулы моносахарида и связаны друг с другом гликозидной связью.
СТРОЕНИЕ МОНОСАХАРИДОВ
Строение простейших оксиальдегидов и оксикетонов с двумя и тремя атомами углерода вытекает непосредственно из их свойств и способов образования.
ОЛИГОСАХАРИДЫ
Олигосахариды обладают, как правило, сладким вкусом; при гидролизе каждая молекула олигосахарида распадается на небольшое число молекул моносахарида (от двух до шести). Общеизвестным примером олигосахаридов является тростниковый, или свекловичный, сахар, гидролизующийся с образованием одной молекулы глюкозы и одной молекулы фруктозы.
В зависимости от числа молекул простых сахаров, образующихся при гидролизе молекулы олигосахарида, различают:
Дисахариды (или биозы), дающие при гидролизе 2 молекулы моносахарида (могут быть одинаковыми и разными):
-сахароза (тростниковый или свекловичный сахар) – при гидролизе распадается на глюкозу и фруктозу;
-лактоза (молочный сахар) – гидролизуется с образованием галактозы и глюкозы;
-мальтоза (солодовый сахар) – распадается на две молекулы d – глюкозы;
Трисахариды (или триозы), дающие при гидролизе 3 молекулы моносахарида ( рафиноза – остаток мальтозы + остаток фруктозы), мелицитоза (остаток сахарозы+ остаток глюкозы) , генцианоза ( остаток мальтозы + фруктозы);
Тетра сахариды (или тетрозы), дающие при гидролизе 4 молекулы моносахарида;
Пентасахариды (или пентозы), дающие при гидролизе 5 молекулы моносахарида и т.д.
Наибольшее значение имеют дисахариды (биозы), т.к чем больше молекулярная масса углеводов, тем менее растворимое вещество и не сладкое на вкус.
Высшие полиозы (высшие полисахариды) сладким вкусом не обладают; при гидролизе каждая молекула полиозы распадается на очень большое число молекул моноз, исчисляемых иногда десятками и сотнями тысяч. Самыми важными представителями высших полиоз, широко распространенными в природе, являются крахмал и целлюлоза.
Полисахариды в природе составляют главную массу орг. в-ва, находящегося в биосфере Земли. Они выполняют в живых организмах три важнейших типа биол. ф-ций, выступая в роли энергетич. резерва, структурных компонентов клеток и тканей или же защитных в-в.
Хорошо известными резервными полисахаридами являются крахмал, гликоген, фруктаны, галактоманнаны и нек-рые р-глюканы. Эти полисахариды способны быстро гидролизоваться имеющимися в клетках ферментами, и их содержание сильно зависит от условий существования и стадии развития организма.
Структурные полисахариды можно разделить на два класса. К первому относят нерастворимые в воде полимеры, образующие волокнистые структуры и служащие армирующим материалом клеточной стенки (целлюлоза высших растений и нек-рых водорослей, хитин грибов, b-D-ксиланы и b-D-ман-наны нек-рых водорослей и высших растений). Ко второму классу относят гелеобразующие полисахариды, обеспечивающие эластичность клеточных стенок и адгезию клеток в тканях. Характерными представителями этого класса полисахаридов являются сульфатир. гликозаминогликаны (мукополисахариды) соединит. ткани животных, сульфатир. галактаны красных водорослей, альгиновые к-ты, пектины и нек-рые гемицеллюло-зы высших растений.
К защитным полисахаридам относят камеди высших растений (гетеро-полисахариды сложного состава и строения), образующиеся в ответ на повреждение растит. тканей, и многочисл. внеклеточные полисахариды микроорганизмов и водорослей, образующие защитную капсулу или модифицирующие св-ва среды обитания клеток.
Вторая ступень классификации связана с расположением карбонильной группы в углеводородной цепи. Характерной особенностью класса углеводов является наличие не менее двух гидроксильных групп и одной карбонильной (альдегидной 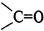 или
или  кетонной) группы. Следовательно, простейший углевод должен содержать три атома углерода. По числу атомов углерода моносахариды называют триозами, тетрозами, пентозами, гексозами и т.д.
кетонной) группы. Следовательно, простейший углевод должен содержать три атома углерода. По числу атомов углерода моносахариды называют триозами, тетрозами, пентозами, гексозами и т.д.
С учетом первой ступени классификации, в названии моноз учитывается как число атомов углерода, так и наличие альдегидной или кетонной группы.
Например, моносахариды, в состав которых входят 6 атомов углерода и альдегидная группа, называются альдогексозами, если же они содержат кето-группу, то кетогексозами.
Следующий этап подразделения углеводов определяется пространственным структурным фактором, а именно, наличием асимметрических центров.
Выделенные звездочкой атомы углерода являются асимметрическими.
Асимметрическим называется атом углерода, соединенный с четырьмя разными заместителями (атомами или группами атомов).
Наличие в молекуле асимметрических атомов углерода делает моносахариды оптически активными соединениями, причем величина удельного вращения является характеристическим параметром моносахарида.
Углеводы, в составе которых есть асимметрические атомы углерода, обладают особым видом пространственной изомерии – стереоизомерией или оптической изомерией. Стереоизомеры отличаются пространственной конфигурацией атомов водорода и гидроксильной группы при асимметрическом атоме углерода. Число стереоизомеров равно 2n, где n - число асимметрических атомов углерода.
Например, альдогексоза общей формулы С6Н12О6 с четырьмя асимметрическими атомами может быть представлена любым из 16 возможных стереоизомеров, восемь из которых относятся к D-ряду, а восемь - к L-ряду.
Приведенные выше линейные структурные формулы альдоз и кетоз называются формулами в проекции Фишера.
Для установления конфигурации углевода следует изобразить его проекцию Фишера в таком виде, чтобы группы СНО и СН2ОН занимали соответственно верхнее и нижнее положения на вертикали.
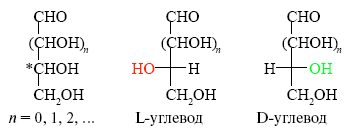
Углевод относятся к L – ряду, если гидроксильная группа при нижнем асимметрическом центре стоит слева, и к D – ряду, если она находится справа от вертикали.
Исторически сложилось так, что глицериновый альдегид, лег в основу D, L – системы для углеводов. В отличие от двух предыдущих примеров L – глицериновый альдегид оказался левовращающим, а его D – форма – правовращающей.
Как видно на слайдах 8,9, молекула альдозы содержит (n-2) асимметрических центра, молекула кетозы – (n-3) таких центра: это значит, что альдогексозы могут существовать в виде 24 (16), конфигурационных изомеров кетогексозы - в виде 23 (8) конфигурационных изомеров.
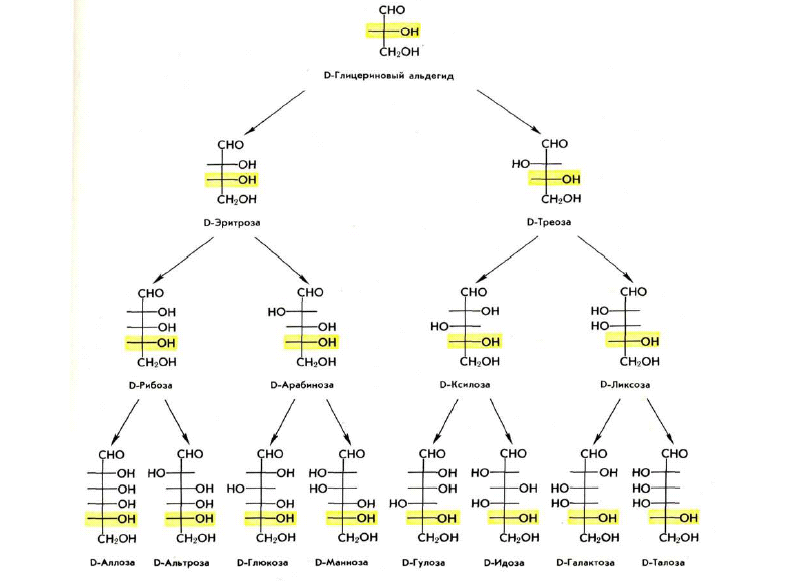
Иерархическая схема стереохимического родства природных альдоз D-ряда
Для перехода от моносахарида D-ряда к L – ряду необходимо изменить на противоположную конфигурацию всех ассиметрических углеродных атомов.
Иерархическая схема стереохимического родства природных кетоз D-ряда
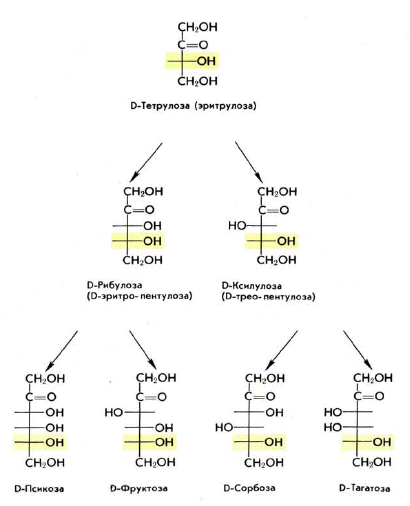
Если принять за минимальную альдозу глицериновый альдегид и минимальную кетозу - эритрулозу, мы можем построить иерархическую схему стереохимического родства природных углеводов с учетом того факта, что, в основном, они относятся к D-ряду в проекциях Фишера.
По мере необходимости указывая в скобках знак оптического вращения: (+) – правовращающий или (-) – левовращающий. Следует заметить, что не существует какой-либо связи между знаком вращения оптического изомера и его принадлежностью к D - , так и L – формы могут быть как лево-, так и правовращающими изомерами и наоборот.
Доказательство строения и стереохимического родства восьми D - гексоз и четырех D-пентоз было основано на блестящих работах Эмиля Фишера, за которые он получил Нобелевскую премию по химии в 1901 году.
Относительно особенностей конфигураций D - и L-рядов моносахаров следует отметить два факта:
1. Обозначение D - и L - не находятся во взаимосвязи со знаком их оптического вращения.
2. Живые организмы "не узнают" и "не умеют" усваивать углеводы L-ряда, тогда как синтезировать они их могут. Свойство инертности ферментов к L - сахарам природа часто использует для построения сложных устойчивых молекулярных систем.
Углеводы были открыты до того, как стали понятны молекулярные аспекты химии.
Вначале было найдено, что они содержат только углерод, водород и кислород (причем два последних в том же соотношении, что и в воде), т. е. являются как бы соединениями угля с водой, поэтому им приписали общую формулу Cx(H2O)y, где х и у — целые числа. Позже стало ясно, что такое определение не является точным, так как были выделены другие соединения, которые по химическим и физическим свойствам хотя и подходят к классу углеводов, однако не содержат водорода и кислорода в соотношении 2:1.
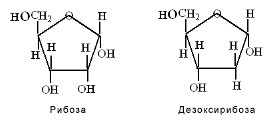
Примером таких соединений может служить дезоксирибоза (входит в состав ДНК), имеющая общую формулу C6H10O4. В данном соединении содержание водорода и кислорода не соответствовало отношению 2:1, но оно тем не менее, имеет свойства углеводов.
Дезоксирибоза. Этот углевод в свободном виде встречается редко. Чаще он входят в состав сложных веществ, т. е. используются в организме в пластических процессах. Дезоксирибоза участвует в биосинтезе дезоксирибонуклеотидов, которые являются структурным компонентом ДНК. Спирт рибитол, производное рибозы, входит в состав витамина В12 и некоторых дыхательных ферментов.
Дезоксирибоза является альдозой. В молекуле дезоксирибозы отсутствует атом кислорода при втором атоме углерода.
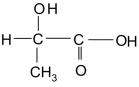
С другой стороны, молочная кислота имеет общую формулу C2H6O3 (оксикарбоновая кислота), однако не принадлежит к классу углеводов.
Позже были открыты углеводы, которые кроме углерода, водорода и кислорода содержали еще азот и серу.
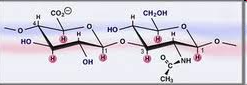
Например гиалуроновая кислота.
К настоящему времени известно около семидесяти моносахаридов, двадцать из которых встречаются в природе, а остальные получены синтетически; но только некоторые из распространенных в природе сахаров представляют для нас интерес. Всем им присущ сладкий вкус, хорошая растворимость в воде, плохая растворимость (или даже нерастворимость) в органических растворителях, например в спирте или эфире.
КОНТРОЛЬНЫЕ ВОПРОСЫ И ЗАДАНИЯ
- Охарактеризуйте углеводы и их биологическую роль в организме.
- Какое свойство углеводов лежит в основе их классификации на моно- ,олиго- и полисахариды. Приведите примеры в виде структурных формул.
- Как образуется циклическая форма моносахаридов? В чем преимущество таких углеводов перед линейными?
- Напишите формулы глюкозы, фруктозы и глицеринового альдегида, а также их фосфорные эфиры.
- Как построены основные дисахариды? Назовите ферменты их гидролиза.
- В чем отличие строения крахмала и гликогена?
- Каковы особенности гидролиза углеводов в процессе пищеварения и их всасывания? Приведите схему гидролиза важнейших углеводов пищи. Какие ферменты ускоряют этот процесс в пищеварительной системе? Какие условия необходимы для действия этих ферментов?
- Каковы механизмы поддержания постоянной концентрации глюкозы в крови?
- Что такое гликемический индекс? Назовите примеры продуктов с низким, средним и высоким гликемическим индексом.
Лекция 7
УГЛЕВОДЫ
Длительное время в науке существовало представление, что моносахариды являются соединениями только с открытой углеродной цепью. Представленные углеводы написаны в проекции Фишера.
Она точно отражает относительную конфигурацию ассиметрических центров, но ничего не говорит (хуже того, говорит весьма искаженно) об истинном расположении атомов в пространстве.
По правилам фишеровской проекции, тетраэдрический углеродный атом располагается так, чтобы его четыре связи проектировались на плоскость в виде креста, причем связи, смотрящие на наблюдателя (над плоскостью бумаги), образуют горизонтальную линию, а уходящие под плоскость бумаги (от наблюдателя) – вертикальную.
В кристаллическом состоянии в природе моносахариды существуют в циклической пираноподобной и фураноподобной формах. Названия циклов происходят от названий родственных гетероциклических соединений – фурана и пирана.
В растворах имеют место взаимные превращения открытой и циклической форм. Циклические формы моносахаридов по химической природе являются циклическими полуацеталями.
Однако ряд экспериментальных фактов не находит объяснения в рамках такого строения моносахаридов.
|
Функциональные группы |
Реагенты |
Примеры качественных реакций на функциональные группы |
Влияние реагентов на исследуемое вещество |
|
активные металлы |
CH3 – CH2 – OH + Na CH3 – CH2 – ONa + H2 |
+ (спец. усл.) |
|
|
гидроксид меди (II) |
+ |
||
|
1) реакция «серебряного зеркала», 2) реакция с фуксинсернистой кислотой |
1) + 2) – |
||
|
лакмус |
- (свеж. пригот.р-р) |
При более глубоком изучении их строения было установлено, что цепные формулы не объясняют некоторых химических свойств углеводов:
1. Почему моносахариды не присоединяют натрия бисульфит (NaHSO2)?
2. Почему они не дают окрашивания с фуксинсернистой кислотой?
3. Чем обусловлены превращения маннозы, глюкозы и фруктозы друг в друга под действием щелочей?
4. Почему у моносахаридов появляется мутаротация, которая связана с изменением угла вращения при стоянии свежеприготовленных растворов?
Если приготовить раствор глюкозы и поместить в поляриметр, то угол вращения равен 113°.
Глюкоза отклоняет плоскость поляризованного луча света на 113°, через 2 часа—на 90°, через 3 часа — на 60° и окончательно — на 52,5°.
5. Почему при рассмотрении формулы глюкозы в ней можно насчитать пять гидроксильных групп и только одна отличается своей реакционной способностью?
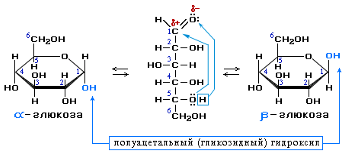
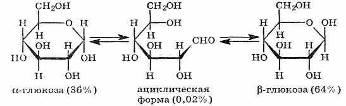
Все эти факты нашли объяснение, когда предположили, что каждый моносахарид может существовать в виде нескольких таутомерных форм. В растворе, кроме развернутых цепей, существуют и циклические формы, которые образуются при внутримолекулярном взаимодействии альдегидной группы и гидроксильной группы при пятом атоме углерода.
Насыщенные шестичленные циклы (в нашем случае пиранозы) на самом деле не плоские. Чтобы валентные углы атомов, входящих в цикл мало отклонялись от наиболее энергетически выгодных – тетраэдрических – углов (109°28’), молекула вынуждена принимать форму зигзага, замкнутого в цикл.
1) в растворах преобладают циклические формы моносахаридов, открытые формы находятся в небольших количествах;
2) изменение оптической активности связано с установлением равновесия между открытой и циклической формами.
Схема таутомерных превращений D-глюкозы
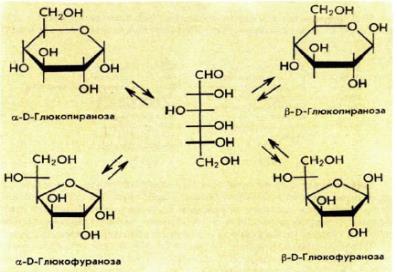
Поэтому углевод в циклической форме (форме Хеуорса) взаимодействует со спиртами с образованием гликозидов. Хеуорс предложил изображать циклические формы сахаров так, чтобы отчетливо были видны и кольцо, и заместители.
Циклические формы моносахаридов могут содержать пять или шесть атомов в цикле. Сахара с шестичленным циклом называются пиранозами, например, глюкоза — глюкопираноза; циклические формы сахаров с пятичленным циклом называются фуранозами. Глюкоза с пятичленным циклом — глюкофураноза, а фруктоза с пятичленным циклом — фруктофураноза.
Циклические формы моносахаридов по химической природе являются циклическими полуацеталями, которые образуются при взаимодействии альдегидной (или кетонной) группы со спиртовой группой моносахарида. В результате внутримолекулярного взаимодействия (АN механизм) электрофильный атом углерода карбонильной группы атакуется нуклеофильным атомом кислорода гидроксильной группы. Образуются термодинамически более устойчивые пятичленные (фуранозные) и шестичленные (пиранозные) циклы. Образование этих циклов связано со способностью углеродных цепей моносахаридов принимать клешневидную конформацию.
В этих реакциях С1 атом из прохирального, в результате циклизации, становится асимметрическим (аномерный центр).
Группа ОН, образовавшаяся на месте альдегидной группы называется полуацетальной или гликозидной. По свойствам она значительно отличается от остальных спиртовых групп моносахарида.
Образование дополнительного хирального центра приводит к возникновению новых стереоизомерных (аномерных) a- и b-форм.
a-Аномерной формой называется такая, у которой полуацетальный гидроксил находится с той же стороны, что и гидроксил у последнего хирального центра, а b-формой - когда полуацетальный гидроксил находится по другую сторону, чем гидроксил у последнего хирального центра.
Образуется 5 взаимно друг в друга переходящих таутомерных форм глюкозы.
Такой вид таутомерии называется циклоцепной или оксо-окси – таутомерией. Таутомерные формы глюкозы находятся в растворе в состоянии равновесия.
В растворах моносахаридов преобладает циклическая полуацетальная форма (99,99%) как более термодинамически выгодная.
Таким образом, моносахариды - циклические полуацетали альдегидо- или кетоно – многоатомных спиртов, существующие в растворе в равновесии со своими таутомерными ациклическими формами.
У свежеприготовленных растворов моносахаридов наблюдается явление мутаротации – изменение во времени угла вращения плоскости поляризации света.
Аномерные a- и b-формы имеют различный угол вращения плоскости поляризованного света.
Так, кристалллическая a, D-глюкопираноза при растворении ее в воде имеет начальный угол вращения +112,5°, а затем он постепенно уменьшается до +52,5°.
Если растворить b, D-глюкопиранозу, ее начальный угол вращения + 19,3°, а затем он увеличивается до +52,5°. Это объясняется тем, что в течение некоторого времени устанавливается равновесие между a- и b – формами: 2/3 b – формы 1/3 a-формы.
Т.о. на долю ациклической формы, содержащей альдегидную группу, приходится менее 0,01%, в связи с этим: 1) - не идет реакция с NaHSO3, 2) - реакция с фуксинсернистой кислотой,
3) - а спектры поглощения растворов глюкозы не показывают наличия полосы, характерной для альдегидной группы.
Насыщенные шестичленные циклы (в нашем случае пиранозы) на самом деле не плоские. Чтобы валентные углы атомов, входящих в цикл мало отклонялись от наиболее энергетически выгодных – тетраэдрических – углов (109°28’), молекула вынуждена принимать форму зигзага, замкнутого в цикл. Есть два типа таких зигзагов, обеспечивающих строго тетраэдрические углы между связями атомов в шестичленном цикле. Один из них, отвечающий обычно минимуму энергии (и, следовательно, реализующийся предпочтительно), называется креслом. Для глюкозы такое кресло, выполненное в духе проекции Хеуорса, представлено формулой «А».
Эта формула очень близка к реальному расположению атомов глюкозы в реальном пространстве, и потому весьма богата информацией. Что же можно в ней усмотреть в первую очередь? Три атома цикла расположены над, а три других – под средней плоскостью, проходящей через центры всех шести связей, образующих цикл. Эта плоскость – основа геометрии кресла. Все пять CH-связей в цисле ориентированы перпендикулярно этой плоскости: параллельно оси цикла и друг другу.
Такие заместители называют аксиальными (от слова axis- ось). Все же гидроксильные группы глюкозы и CH2OH-группа расположены вблизи плоскости цикла (точнее, их связи с циклом образуют с этой плоскостью углы около 30°). Такие заместители называют экваториальными. Теперь видно, что пиранозный цикл в глюкозе охвачен, именно по экватору, почти равномерным кольцом из гидроксильных групп. А в галактозе, например, эта равномерность уже нарушена: гидроксил при С-4 занимает аксиальное положение и его химические свойства существенно отличны от свойств гидроксила при С-4 в глюкозе и всех остальных гидроксилов в галактозе (см. формулу «Б»).
Впервые предположение о циклическом строении глюкозы было высказано русским ученым А.А. Колли (1870), а затем развито немецким ученым Б. Толленсом (1883).
ХИМИЧЕСКИЕ СВОЙСТВА УГЛЕВОДОВ
1 Реакции моносахаридов с открытой углеродной цепью
Химические свойства моносахаридов обусловлены наличием в их молекулах различных функциональных групп.
Глюкоза легко окисляется. В зависимости от характера окислителей получаются различные продукты.
1) Окисление под действием слабых (мягких) окислителей с образованием глюконовой кислоты.
К числу таких реакций относятся качественные реакции на глюкозу как альдегид:
а) реакция с аммиачным раствором оксида серебра (реакция «серебряного зеркала»).
Реакции моносахаридов с открытой углеродной цепью
1 Окисление моносахаридов мягкими окислителями дает альдоновые кислоты:
а)взаимодействие гидроксид иона с диамином серебра:
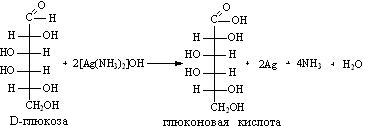
В результате получается глюконовая кислота, при взаимодействии которой с углекислым кальцием получается глюконат кальция – известное лекарственное средство.
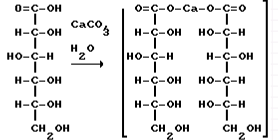
Глюконовая кислота Глюконат кальция
б) реакция с гидроксидом меди (II) в щелочной среде при нагревании. Происходит восстановлении иона двухвалентной меди до одновалентной за счёт окисления альдегидных групп.
С6Н12О6+2 Cu SO4+5 NaOH C6H11O6Na + 2CuOH +2Na2SO4 + 2H2O
Cu2O + H2O
Реакция Троммера
При этом на холоде выпадает оранжево-жёлтый осадок гидрата закиси меди СuОН, а при нагревании – красный осадок закиси меди Сu2O.
2. Реакция спиртовых гидроксидов:
а) Взаимодействие с гидроксидом меди (II) с образованием алкоголята меди (II):
C6H12O6 + Cu(OH)2 > C6H10O6Cu + H2O
в) образование сложных эфиров при взаимодействии с карбоновыми кислотами — реакция этерификации. Например, взаимодействие глюкозы с уксусной кислотой или ее хлорангидридом:
C6H12O6+5CH3COOHC6H7O6(CH3CO)5
3) Окисление под действием сильных окислителей, (например азотной кислоты или пероксид водорода), приводят к образованию двухосновных сахарных кислот (глюкаровой кислоты).
В ходе этой реакции альдегидная – СНО и первичная спиртовая группа –СН2ОН окисляются до карбоксильных групп – СООН.
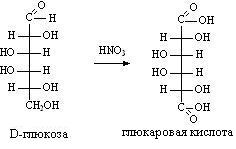
4) Восстановление амальгамой натрия, алюмогидридом лития или боргидридом натрия приводит к образованию шестиатомных спиртов:
Взаимодействие углеводов с карбоновыми кислотами идет с образованием сложных эфиров.
5) реакция этерификации:
взаимодействие с карбоновыми кислотами с образованием сложных эфиров
C6H12O6+5CH3COOHC6H7O6(CH3CO)5
6) Образование оксимов
Моносахариды легко реагируют с гидроксиламином NH2OH, дальнейшая дегидратация приводит к нитрилам, которые при отщеплении циановодорода образуют альдозы с меньшим числом атомов углерода. Так можно установить строение моносахарида и его принадлежность к D или L ряду.
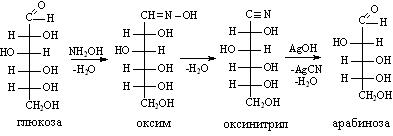
7) Эпимеризация моноз
Эпимеризация моноз происходит под действием щелочей и связана с образованием общегоенола. В результате получается равновесная смесь глюкозы, маннозы и фруктозы:
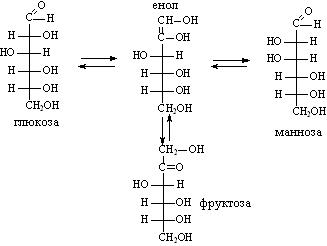
8) Синтез Килина-Фишера - присоединение по карбонильной группе
На этой реакции основан синтез Килиани – Фишера, позволяющий переходить от углеводов с меньшим количеством атомов углерода к углеводам с более длинной углеродной цепью. Для этого полученные нитрилы подвергают гидролизу, а затем восстанавливают амальгамой натрия.
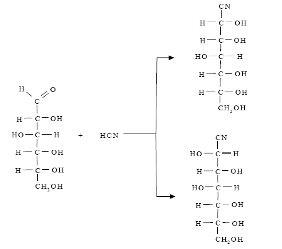
9) Восстановление углеводов (глюкозы)до шестиатомного спирта (сорбита)
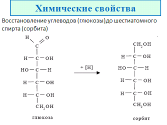
При восстановлении углеводов получают многоатомные спирты – глициты.
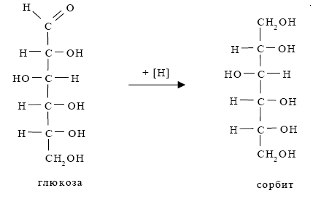
Шестиатомный спирт, полученный при восстановлении глюкозы, называется сорбитом и используется в качестве заменителя сахара для больных сахарным диабетом.
10) Окисление углеводов в присутствии ферментов:

Избирательное окисление первичной спиртовой группы в присутствии ферментов.
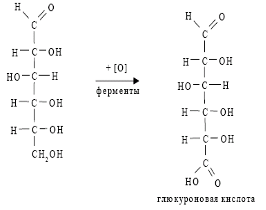
Реакция протекает в организмах животных и человека, но в лабораторных условиях ее осуществить сложно.
Для открытой формы глюкозы характерны все химические реакции альдегидов, которые идут при нагревании, так как при повышенной температуре содержание открытой формы глюкозы достаточно велико.
11) Реакция с фенилгидразином
Реакция с фенилгидразином дает на первой стадии фенилгидразон, затем следующая молекула фенилгидразина окисляет соседний с карбонильным углеродом гидроксил, гидролиз которого дает кетальдегид, а дальнейшее восстановление - кетогексозу:
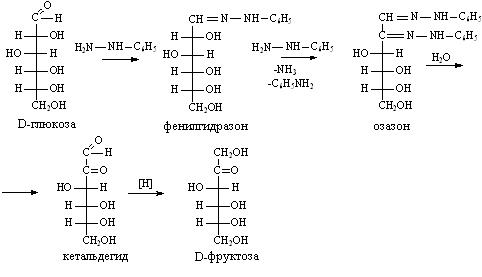
2 Реакция моносахаридов в циклической форме
1 Образование гликозидов
Гликозидный гидроксил легко вступает в реакции со спиртами, аминами, тиозами, образуя O, N или S-гликозиды, например при действии на -D-глюкопиранозу этанола в присутствии соляной кислоты образуется О-этилгликозид -D-глюкопиранозы:
Образующийся гликозид уже не способен к переходу в открытую форму.
2 Алкилирование происходит под действием алкил галогенидов, при этом алкилируются все гидроксилы.
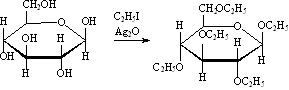
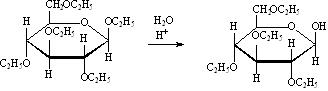
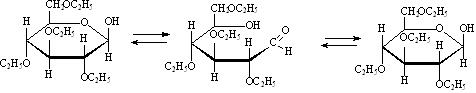
При гидролизе образовавшейся пентаэтил- -D-глюкопиранозы освобождается только гликозидный гидроксил.
В результате получается тетрааэтил- -D-глюкопираноза, наличие свободного гликозидного
гидроксила позволяет ей переходить в открытую форму и, соответственно в тетрааэтил- -D- .
глюкопиранозу
3. Ацилирование под действием галоген ангидридов или ангидридов кислот приводит к образованию ацильных производных, например при ацетилировании -D-глюкопиранозы образуется пентаацетил- -D-глюкопираноза:
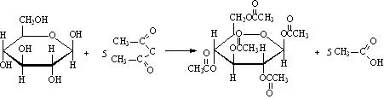
4. Образование хелатных комплексов.
Как многоатомные спирты моносахориды при действии гидроксидов переходных металлов, например гидроксида меди (II), образуют растворимые комплексы. Комплексообразование происходит за счет кислородов гидроксильных групп:
Важнейшим свойством моносахаридов является их ферментативное брожение, т.е. распад молекул на осколки под действием различных ферментов. Брожению подвергаются в основном гексозы в присутствии ферментов, выделяемых дрожжевыми грибками, бактериями или плесневыми грибками.
В зависимости от природы действующего фермента различают реакции следующих видов:
а) спиртовое брожение C6H12O6 2C2H5OH (этанол)+ 2CO2
б) молочно-кислое брожение C6H12O6 2CH3-CH(OH)-COOH
молочная кислота
в) масляно-кислое брожение C6H12O6 C3H7COOH + 2CO2 + 2H2O
масляная кислота
г) лимонно-кислое брожение
C6H12O6 + O2 HOOC-CH2-C(OH)(COOH)-CH2-COOH + 2H2O
лимонная кислота
д) ацетон-бутанольное брожение
2C6H12O6 С4H9OH + СH3-СO-CH3 + 5CO2 + 4H2
бутанол ацетон
В живом организме в процессе метаболизма глюкоза окисляется с выделением большого количества энергии:
C6H12O6 + 6O2 6CO2 + 6H2O + 2920 кДж
Лекция 8
(продолжение 7)
УГЛЕВОДЫ
Все полисахариды построены по типу гликозидов. При их образовании выделяется вода, как правило за счет полуацетального гидроксила одной молекулы и какого-либо гидроксила (полуацетального или обычного спиртового) другой молекулы, например:
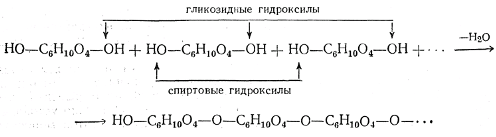
При образовании дисахарида одна молекула моносахарида всегда образует связь со второй молекулой с помощью своего полуацетального гидроксила. Что касается второй молекулы моносахарида, то она может участвовать в образовании этой связи либо также своим полуацетальным гидроксилом, либо каким-либо из остальных, т.е. спиртовых, гидроксилов; в последнем случае один палуацетальный гидроксил в молекуле дисахарида будет оставаться свободным.
Олигосахариды (до 10 молекул моносахаров).
Наиболее распространенными в природе олигосахаридами являются дисахариды.
|
Олигосахариды
- Мальтоза -Лактоза
|
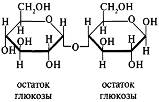
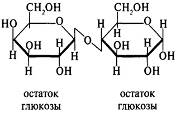
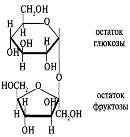 Сахароза
СахарозаМальтоза образуется из полисахаридов как промежуточный продукт. Она состоит из двух остатков глюкозы, соединенных между собой -1,4-гликозидной связью.
Лактоза содержится в молоке животных и человека. В состав лактозы входит остаток галактозы и глюкозы; эти монозы связаны между собой -1,4-гликозидной связью.
Сахароза - наиболее распространенный и важный дисахарид, встречающийся в растительном мире. Сахароза является ценным питательным веществом для человека. Сахароза состоит из остатков -D-глюкозы и -D-фруктозы, связанных , -1,2-гликозидной связью.
Полисахариды представляют собой биополимеры, мономерами которых служат моносахариды. Если в составе полисахарида содержатся остатки моносахарида одного вида, его называют гомополисахаридом, если разных, - гетерополисахаридом. По своему функциональному назначению гомополисахариды могут быть разделены на две группы: структурные и резервные полисахариды. Важным структурным гомополисахаридом является целлюлоза, а главным резервным – гликоген и крахмал (у животных и растений соответственно)
Крахмал - гомополисахарид, состоящий из остатков глюкозы. Он является одним из наиболее распространенных запасных полисахаридов растений. Крахмал накапливается в семенах, клубнях (40 - 78%) и других частях растений (10 - 25%). Крахмал состоит из двух фракций, отличающихся строением и свойствами: амилозы - 15 - 25% и амилопектина - 75 - 85%.
Фрагмент молекулы крахмала
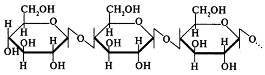
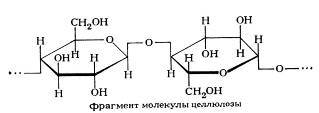
Оба полимера построены в основном из остатков глюкозы, связанных в положении (14).
Амилоза построена из остатков глюкозы, связанных кислородными "мостиками" (гликозидными связями) между первым атомом углерода одного остатка и четвертым углеродным атомом другого:
Глюкозные остатки образуют неразветвленную цепь с молекулярной массой от 16 до 160 кДа. Эта цепь в пространстве закручивается в спираль, но молекула в целом имеет нитевидную форму.
К физиологически важным гомополисахаридам относят крахмал и гликоген. К числу важнейших гетерополисахаридов - гиалуроновую кислоту, хондротинсульфат и гепарин.
Две молекулы -глюкозы связываются с образованием мальтозы - солодового сахара (упрощенная формула):
Целлюлоза - наиболее распространенное органическое соединение. Она встречается в растительном мире в качестве структурного компонента клеточной стенки. Особенно богаты целлюлозой волокна хлопка (98 - 99%). Целлюлоза состоит из остатков глюкозы, связанных между собой -1,4-гликозидньши связями.
Структура целлюлозы хорошо отвечает ее биологической задаче. Отдельные цепи целлюлозы связаны между собой водородными связями, что способствует образованию волокнистой и очень прочной структуры. В клеточных стенках растений волокна целлюлозы плотно упакованы в слои, которые дополнительно стабилизированы другими соединениями полисахаридной природы.
Целлюлоза не имеет питательной ценности для высших животных и человека, так как пищеварительные секреты слюны и ферменты желудочно-кишечного тракта не способны расщеплять 1,4-гликозидные связи до глюкозы.
Биологические функции полисахаридов
Энергетическая - крахмал и гликоген составляют "депо" углеводов в клетке; при необходимости они быстро расщепляются на легко усваиваемый источник энергии - глюкозу.
Опорная - хондроитинсульфат выполняет опорную функцию в костной ткани.
Структурная - гиалуроновая кислота, хондроитинсульфат и гепарин являются структурными межклеточными веществами.
Гидроосмотическая и ионрегулирующая - гиалуроновая кислота, благодаря высокой гидрофильности и отрицательному заряду, связывает межклеточную воду и катионы, регулируя межклеточное осмотическое давление.
Молекулы целлюлозы отличаются от крахмала, имеющего такую же брутто-формулу, более высокой степенью полимеризации. Последняя у целлюлозы составляет 2500-3000 и иногда доходит даже до 4000, тогда как у крахмала она находится в пределах 600-900. Кроме того, целлюлоза построена из звеньев -глюкозы, а крахмал - из -глюкозы. Указанные формы глюкозы очень мало отличаются друг от друга своим пространственным строением: (рис.1). Однако, как ни мало это различие, оно сказывается на строении полимеров и на их свойствах.
2) Поли – (>10 молекул)
|
Гликоген
|
Хитин
|
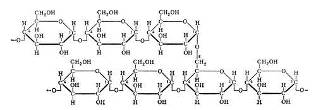
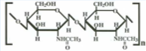
Хитин – главный скелетный полисахарид беспозвоночных и компонент клеточной стенки грибов и некоторых зеленых водорослей. В кутикуле членистоногих образует комплексы с белками, пигментами, солями кальция. Длинные параллельные цепи хитина также, как и цепи целлюлозы, собраны в пучки. По своей структуре хитин очень близок к целлюлозе, за одним исключением: при втором атоме углерода гидроксильная группа ОН заменена группой NН – СО–СН3. Получают хитин обработкой исходного материала (обычно панцирей ракообразных) кислотами, щелочами или окислителями. Молекулярная масса выделенного хитина 151 –200 тыс. При обработке хитина щелочами в жестких условиях происходит N- дезацетилирование с образованием хитозана (растворимого в кислотах), который применяется в производстве бумаги и для удаления примесей из водных растворов. целлюлозахитин
К физиологически важным гомополисахаридам относят и гликоген.

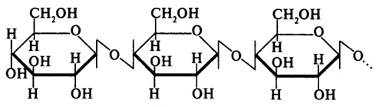 Крахмал
Крахмал
В качестве примера разветвленного гомогликана здесь представлен фрагмент молекулы гликогена.
B гликогене точки ветвления располагаются в среднем через каждые 8-10 остатков глюкозы.
Связи в точках ветвления находятся в положении (18), остальные остатки боковой цепи связаны в положении (14). За счет этого образуется разветвленная, древовидная структура, в которой имеется только одна аномерная ОН-группа, т.е. только один восстанавливающий конец.
В качестве примера разветвленного гомогликана здесь представлен фрагмент молекулы гликогена. Похожее строение имеет амилопектин, разветвленный компонент растительного крахмала.
Крахмал - гомополисахарид, состоящий из остатков глюкозы. Он является одним из наиболее распространенных запасных полисахаридов растений. Крахмал накапливается в семенах, клубнях (40 - 78%) и других частях растений (10 - 25%). Крахмал состоит из двух фракций, отличающихся строением и свойствами: амилозы - 15 - 25% и амилопектина - 75 - 85%.
Оба полимера построены в основном из остатков глюкозы, связанных в положении (14). B гликогене точки ветвления располагаются в среднем через каждые 8-10 остатков глюкозы. Связи в точках ветвления находятся в положении (18), остальные остатки боковой цепи связаны в положении (14). За счет этого образуется разветвленная, древовидная структура, в которой имеется только одна аномерная ОН-группа, т.е. только один восстанавливающий конец.
Крахмал и гликоген относятся к физиологически важным поли-сахаридам.
Амилоза построена из остатков глюкозы, связанных кислородными "мостиками" (гликозидными связями) между первым атомом углерода одного остатка и четвертым углеродным атомом другого:
Глюкозные остатки образуют неразветвленную цепь с молекулярной массой от 16 до 160 кДа. Эта цепь в пространстве закручивается в спираль (рис. 12), но молекула в целом имеет нитевидную форму.
Сложную структуру имеет линейный гетерогликан муреин, который в качестве структурного полисахарида придает прочность клеточным стенкам бактерий.
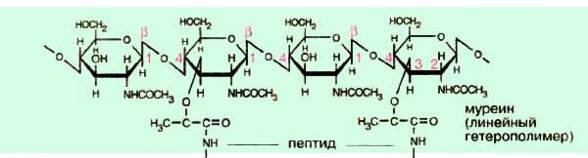
На схеме приведен только один сегмент этой нитевидной молекулы.
В муреине чередуются остатки двух различных моносахаридов, связанных в положении (14): N-ацетилглюкозамина (GlcNAc) и характерной для муреина N-ацетилмурамовой кислоты (MurNAc).
Последняя является простым эфиром молочной кислоты с N-ацетилглюкозамином.
В клеточной стенке карбоксильная группа молочной кислоты связана амидной связью с пептидом (на схеме показан условно), который соединяет отдельные цепи муреина в трехмерную сетчатую структуру (на схеме не приведена).
Глюкозные остатки образуют неразветвленную цепь с молекулярной массой от 16 до 160 кДа. Эта цепь в пространстве закручивается в спираль (рис. 12), но молекула в целом имеет нитевидную форму.
|
Спиральная конформация молекулы амилозы |
|
 Амилоза
Амилоза
 Амилопектин
АмилопектинАмилопектин имеет молекулы с разветвленной цепью остатков глюкозы, образованной за счет связи между шестым атомом углерода одного остатка и первым углеродным атомом другого:
Гликоген - резервное питательное вещество организма человека и животных. Иначе его называют "животным крахмалом". В организме человека он накапливается в печени (-20%) и в мышцах (-2%). Гликоген по структуре близок к амилопектину, однако степень ветвления у него больше, чем у амилопектина, поэтому молекула гликогена более компактна. Гликоген - не однородное вещество, а представляет собой смесь полисахаридов разной молекулярной массы. Часть его находится в соединении с белками.
Гепарин - гетерополисахарид, препятствующий свертыванию крови у животных и человека. Гепарин содержится в крови, печени, легких, селезенке, щитовидной железе и в других тканях и органах.
Молекула гепарина состоит из глюкуроновой кислоты и -глюкозамина в виде двойного сульфопроизводного, соединенных между собой -1,4-гликозидными связями.
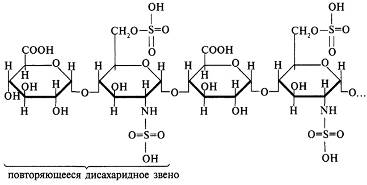
Лекция 8
1 ЛИПИДЫ
Липиды - это гетерогенная группа веществ, в состав которых входят жирные кислоты.
Для всех липидов характерны свойства гидрофобности и липофильности, это значит, что липиды плохо растворимы в воде и хорошо растворимы в неполярных растворителях (спирты – фенол, стирол, эфир, хлорофол, толуол, бензол). Липиды делятся на три группы в зависимости от сложности строения: 1) простые липиды, 2) сложные липиды.
Жиры - это природные соединения, которые представляют собой сложные эфиры - (*- общая формула сложных эфиров) - глицерина и высших карбоновых кислот: где R, R' и R'' – углеводородные радикалы высших карбоновых (жирных) кислот, преимущественно от С3 до С17. Карбоновые кислоты могут быть различными, но всегда нормального строения и, как правило, с четным числом атомов углерода.
Строение жиров:
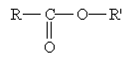
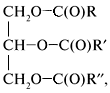
Название:
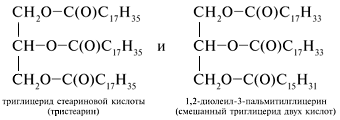
|
Триглицерид стеариновой кислоты (тристеарин) |
1,2-диолеил-3-пальмитилглицерин (смешанный триглицерид двух кислот) |
Простой жир Смешанный жир
Т.О. мы говорим – жир, а на самом деле это – эфир! Эфир кислот и глицерина.
Жиры бывают “простыми” и “смешанными”.
В состав простых жиров входят остатки одинаковых кислот (т. е. R’ = R" = R‘ - триглицерид стеариновой кислоты - ТРИСТЕАРИН),
в состав смешанных — остатки различных кислот (1,2–диолеил–3- пальмитилглицерин [смешанный триглицерил двух кислот – ОЛЕИНОВОЙ и ПАЛЬМИТИЛОВОЙ].
Жиры – главная составная часть жировых клеток животных и растений – являются одним из важнейших пищевых резервов организма. При окислении жиров выделяется значительно больше энергии, чем при окислении углеводов и белков.
Три наиболее распространенные в природе жирные кислоты – это предельные кислоты: пальмитиновая (С16), стеариновая (С18) – и непредельная олеиновая (С18) кислота. У большинства ненасыщенных жирных кислот, входящих в состав жиров, масел и биологических мембран, преобладающим является цисизомер, трансизомер встречается редко. Чем выше степень ненасыщенности жирной кислоты, тем ниже ее температура плавления (табл).
Также жирах наиболее часто встречаются следующие жирные кислоты:
) Алкановые кислоты (С2Н2n-1)
1. Масляная кислота СН3 — (CH2)2 — СООН
2. Капроновая кислота СН3 — (CH2)4 — СООН
3. Пальмитиновая кислота СН3 — (CH2)14 — СООН
4. Стеариновая кислота СН3 — (CH2)16 — СООН
) Алкеновые кислоты (С2Н2n-2)
5. Олеиновая кислота С17Н33СООН
СН3—(СН2)7—СН = СН—(СН2)7—СООН
) Алкадиеновые кислоты
6. Линолевая кислота С17Н31СООН
СН3—(СН2)4—СН = СН—СН2—СН = СН—СООН
V) Алкатриеновые кислоты
7. Линоленовая кислота С17Н29СООН
СН3СН2СН = CHCH2CH = CHCH2CH = СН(СН2)4СООН
2 НОМЕНТКЛАТУРА И ИЗОМЕРИЯ ЖИРОВ
Природные жиры (триацилглицерины) являются триэфирами глицерина и жирных кислот. Обычное название этих соединений – триглицериды. Известны не только глицериды одинаковых кислот (простые глицериды), но и преимущественно разных кислот (смешанные глицериды). Например:
Названия сложных эфиров производят от названия углеводородного радикала и названия кислоты, в котором вместо окончания -овая используют суффикс -ат, например:
Для сложных эфиров характерны следующие виды изомерии:
1. Изомерия углеродной цепи начинается по кислотному остатку с бутановой кислоты, по спиртовому остатку — с пропилового спирта, например, этилбутирату изомерны этилизобутират, пропилацетат и изопропилацетат.
2. Изомерия положения сложноэфирной группировки —СО—О—. Этот вид изомерии начинаетсясо сложных эфиров, в молекулах которых содержится не менее 4 атомов углерода, например этилацетат и метилпропионат.
3. Межклассовая изомерия, например, метилацетату изомерна пропановая кислота.
Для сложных эфиров, содержащих непредельную кислоту или непредельный спирт, возможны еще два вида изомерии: изомерия положения кратной связи и цис-, транс-изомерия.
|
Структурная формула |
Название |
Температура плавления, °С |
|
Насыщенные жирные кислоты |
||
|
СН3(СН2)10СООН |
Лауриновая |
44 |
|
Ненасыщенные жирные кислоты |
||
|
СН3(СН2)5СН=СН(СН2)7СООН |
Пальмитин-олеиновая |
–1 |
Жирные кислоты - относятся к группе карбоновых кислот.
Карбоновые кислоты это такие органические кислоты, которые содержат в себе хотя бы одну карбоксильную группу. Классификация карбоновых кислот основана на количестве карбоксильных групп. Жирные кислоты относятся к монокарбоновым кислотам. С точки зрения химического строения все карбоновые кислоты делятся на две группы:
1) насыщенные или предельные карбоновые кислоты, в радикале которых встречаются только одинарные связи между атомами углерода.
2) непредельные или ненасыщенные, в радикале которых, встречаются двойные связи. Количество двойных связей является классификационным признаком, который обозначается суффиксом – ен.
Биологическое значение имеют коротко радикальные предельные кислоты с С1 до С8.такие коротко радикальные кислоты являются важными промежуточными продуктами метаболических путей в клетке.
После С8 биологическое значение имеют только жирные кислоты с четным количеством атомов углерода в радикале, т.к. все они синтезируются на основе уксусной кислоты.
В организме встречаются предельные жирные кислоты до С24, с увеличением длинны радикала, изменяется фазовое состояние кислоты.
Коротко радикальные жирные кислоты являются жидкостями. Чем длиннее радикал, тем тверже кислота.
Среди непредельных жирных кислот биологическое значение имеет тетроеновые, пентоеновые и гексаеновые жирные кислоты.
Пентоеновые и гексаеновые встречаются в рыбьем жире.
Тетроеновая в арахисовом масле.
Степень насыщенности жирной кислоты определяет ее фазовое состояние.
Насыщенные жирные кислоты являются твердыми, ненасыщенные – жидкими. Молекулы жирных кислот сочетают в себе два свойства и гидрофобности и гидрофильности, поэтому говорят, что они обладают амфотерными свойствами. Если радикал жирной кислоты достаточно короткий, то она растворима в воде, если радикал длинный, то плохо растворима в воде.
Простые липиды - это сложные эфиры жирных кислот и спиртов. Они образуются за счет реакции этерификации.

Все простые липиды делятся на три группы:
1) воска; 2) жиры; 3) Церамид
3 ВОСКА
Это сложные эфиры жирной кислоты с одноатомным спиртом. Воска характерны для растительного мира и часто покрывают вегетативные органы растений, живущих в засушливых условиях (каменный плющ, кактусы, брусника). Препятствуют излишнему испарению воды, отражает солнечные лучи, что препятствует перегреванию растений и избыточному ультрафиолетовому облучению. Воска у животных менее распространены, у насекомых восковой налет покрывает кутикулу, препятствуя испарению воды. У человека также встречаются воска, которые выделяются на поверхность эпидермиса и производных эпидермиса, например волосы и ногти.
4 ЖИРЫ
Это сложные эфиры жирных кислот с глицерином.
Агрегатное состояние жира при физиологических температурах зависит от того, какие жирные кислоты входят в его состав. Жиры, которые при физиологических температурах находятся в жидком состоянии, называются маслами. Жиры, которые при физиологических температурах находятся в твердо состоянии, называются жирами.
Возможны различные варианты реакций с глицерином.
С глицерином может прореагировать от 1 до 3 жирных кислот.
- моноацилглицерол (МАГ)
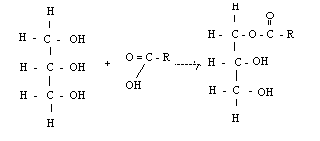
- диацилглицерол (ДАГ)
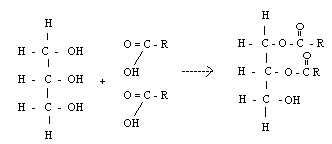
- триацилглицерол (ТАГ)
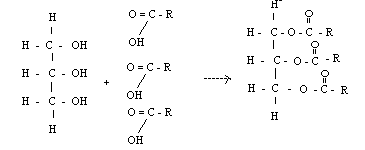
5 ФИЗИЧЕСКИЕ СВОЙСТВА
- Животные жиры плавятся при высокой t0C
- Растительные жиры при низкой t0С
- Высокая вязкость
- Слабо проводят тепло и электричество
- Плохо растворяются в воде
- Растворяются в бензине, бензоле,хлороформе
Температуры плавления индивидуальных триглицеридов, входящих в состав жиров, зависят от длины цепи, степени ненасыщенности жирных кислот и их расположения в триглицеридах.
Подобно большинству длинноцепочных соединений, триглицериды могут кристаллизоваться в несколько полиморфных формах. Полиморфизм проявляют не только индивидуальные триглицериды, но и природные жиры, состоящие из триглицеридов с близкими длинами кислотных цепей.
Жиры, являющиеся смесью различные триглицеридов, не имеют четкой температуры плавления.
Все жиры характеризуются значительным увеличением объема при плавлении.
При постепенном охлаждении жидкий жир частично кристаллизуется и приобретает форму твердого тела, обладающего пластичностью. Пластичность характерна для жиров, содержащих 10-30% кристаллич. фазы. В пластичном жире кристаллы твердых триглицеридов образуют решетку, внутри которой находится значительной количество жидкой фазы. При дальнейшем охлаждении все триглицериды кристаллизуются и жир теряет пластичность. Последняя является ценным свойством жиров, особенно пищевых.
Важная характеристика жира - твердость, определяемая нагрузкой в г/см, необходимой для разрезания жира в определенных условиях.
Жиры обладают низким давлением паров и кипят только в высоком вакууме (~ 250 °С при 0,001 мм рт. ст.).
Плотность жира зависит от молекулярной массы жирных кислот и степени их ненасыщенности и может быть рассчитана по формуле: d1515 = 0,8475 + 0,0003 числа омыления + 0,00014 йодного числа.
Температурный коэффициент объемного расширения жиров 0,0007/К.
Показатель преломления жира зависит от молекулярной массы жирных кислот и степени их ненасыщенности: nD40 = 1,4643; 0,000066 - числа омыления; 0,0096 + 0,0001171 йодного числа.
Температурный коэффициент рефракции жира - 0,0036/К. Из-за сильного межмол. взаимодействие жирно-кислотных цепей вязкость жира высока ( - 2-4 мкПа * с при 40 °С).
Поверхностное натяжение большинства жиров на границе жир - воздух 30-35 мН/м.
Энтальпия сгорания жира (в Дж/г) определяется формулой: — D Hсгор = 47645 - 4,1868 йодного числа - 38,31 числа омыления и для большинства жиров составляет 39,5 кДж/г; D Hпл 120-150 Дж/г; Сор около 2 Дж/(г * К).
Жиры плохие проводники тепла и электричества. Коэф. теплопроводности 0,170 Вт/(м * К), диэлектрическая постоянная (30-40) * 10 - 30 Кл * м.
Температура вспышки большинства жиров 270-330°С, температура самовоспламенения 340-360 °С; характеристикой ЖИРЫ является также так называемой температура дымообразования (дымления), при которой происходит визуально заметное образование дыма вследствие разложения ЖИРЫ Она падает с ростом кислотного числа ЖИРЫ и лежит в пределах 160-230°С. ЖИРЫ неограниченно растворим в диэтиловом эфире, бензоле, хлороформе, частично растворим в этаноле (5-10%) и ацетоне, практически не растворим в воде, но образуют с ней эмульсии. В 100 г воды эмульгируются 10 мг говяжьего жира, 50 мг свиного. Жиры растворяют небольшие количества воды (0,1-0,4%) и значительной кол-ва газов (7-10% по объему N2, H2, О2 и до 100% СО2). Растворимость Н2, N2, O2 возрастает с ростом температуры, растворимость СО2 падает.
6 ХИМИЧЕСКИЕ СВОЙСТВА ЖИРОВ
- гидролиз
- гидрирование
- прогоркание
- омыление
6.1 Гидролиз или омыление, жиров
Под гидролизом жиров подразумевают гидролитическое расщепление глицеридов.
Применяют четыре основных способа гидролиза жиров:
1) Омыление жиров водой ведут под влиянием ферментов или серной кислоты:
Гидролиз липидов, приводящий к образованию плохо пахнущих карбоновых кислот (например, масляной);
1) Прогоркани -перекисное окисление липидов
|
|
|
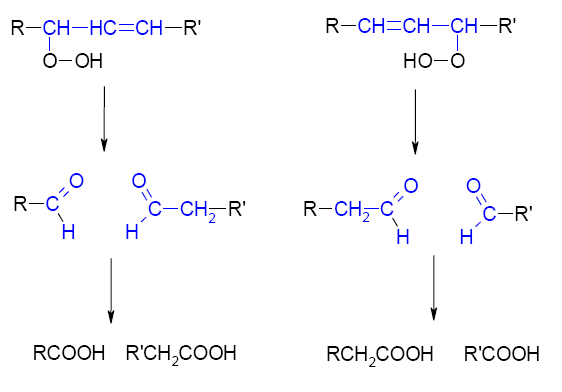
Окисление липидов – является основной причиной повреждения клеточных мембран, например при лучевой болезни. Однако in vivo ПОЛ постоянно идет и без наличия радиактивного излучения. При окислении железа (Fe+2) образуются гидроксильные радикалы (Н-О)– и гидропероксидные радикалы (Н-О-О)–, которые инициируют окисление липидов. Реакция имеет цепной характер и идет с остатками ненасыщенных жирных кислот:
В результате образуются альдегиды и карбоновые кислоты, мембраны повреждаются, а радикальные метаболиты обладают мутагенными и канцерогенным действием. Защитой от окисления липидов являются антиоксиданты – например, витамин Е.
Глицерин, входящий в состав жира, подвергается окислению и дегидратации при нагревании жира с конц. серной кислотой. Ощущается неприятный запах акролеина. Это «акролеиновая проба», позволяющая отличить жиры от жироподобных веществ.
2) Гидролиз жиров в щелочной среде дает глицерин и растворимые соли карбоновых кислот:
3) В результате окисления жиров наряду с освобождением энергии образуется довольно много воды. При недостатке питьевой воды это позволяет легче переносить жажду:
4) Гидрирование жиров – превращение жидких растительных масел в твердые жиры – имеет большое значение для пищевых целей. Оно идет при высокой температуре или высоком давлении в присутствии специальных катализаторов. Так в промышленности получают маргарин.
Слайд 33
Слайд 34
Слайд 35
Слайд 36
Слайд 37 - При щелочном гидролизе фосфолипидов образуются: глицерин, соли карбоновы кислот, фосфат натрия и спирт
Слайд 38 - Количественной характеристикой ненасыщенных липидов служит иодное число, которое соответствует массе йода (в граммах), которая может присоединиться к 100 г липида.
Слайд 40 –
Слайд 42
ХИМИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ Е.В. СЕРЕБРЯКОВА