Синтез гликозидов N-ацетил-D-глюкозамина с агликонами пиразолоизохинолинов с использованием каталитической межфазной системы «твердое тело – органический растворитель» для изучения их медико-биологические свойства
PAGE \* MERGEFORMAT3
ОГЛАВЛЕНИЕ
|
|
Стр
|
|
ПЕРЕЧЕНЬ УСЛОВНЫХ ОБОЗНАЧЕНИЙ
|
4
|
|
ВВЕДЕНИЕ
|
5
|
|
1. МЕТОДЫ СИНТЕЗА ПИРАЗОЛОИЗОХИНОЛИНОВ
|
7
|
|
2. ГЛИКОЗИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
|
16
|
|
3. МАТЕРИАЛЫ И МЕТОДИКА ИССЛЕДОВАНИЯ
|
23
|
|
4. ОХРАНА ТРУДА И БЕЗОПАСНОСТЬ В ЧЕРЕЗВЫЧАЙНЫХ
СИТУАЦИЯХ
|
28
|
|
4.1. Карбонат калия
|
30
|
|
4.2. Ацетонитрил
|
30
|
|
4.3. Хлороформ
|
31
|
|
4.4. Изопропиловый спирт
|
31
|
|
4.5. Ацетон
|
32
|
|
4.6. Бромид ртути (II)
|
32
|
|
4.7. Краун-эфир
|
33
|
|
4.8. Электробезопасность
|
33
|
|
4.9. Техника безопасности при работе с персональным компьютером
|
34
|
|
5. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
|
36
|
|
ВЫВОДЫ
|
39
|
|
СПИСОК ЛИТЕРАТУРЫ
|
40
|
|
ПРИЛОЖЕНИЕ
|
44
|
|
|
|
ПЕРЕЧЕНЬ УСЛОВНЫХ ОБОЗНАЧЕНИЙ
15К5 – 15-Краун-5
Ме – метил
ТБАГ–тетрабутиламмоний гидросульфат
ТГБК – тетрагидро--карболин
ТГИ – тетрагидроимидазопиридин
ТГИХ – тетрагидроизохинолин
ТСХ – тонкослойнаяхроматография
ТЭБАХ – бензилтриэтиламмоний хлорид
ХС – химический сигнал
ЯМР – ядерный магнитный резонанс
LD50 – средняя доза вещества, вызывающая гибель половины членов испытуемой группы
Ph – фенил
ВВЕДЕНИЕ
Производные изохинолина более столетия остаются популярными объектами исследования для различных областей химии и медицины. Фрагмент изохинолина входит в состав изохинолиновых алкалоидов, обладающих физиологической активностью и поэтому представляющих интерес, как лекарственные средства. Например, к производным N-бензилизохинолина относятся гидрохлорид опиумного алкалоида папаверина и его синтетический аналог - дротаверин (но-шпа). Современные исследования на сегодняшний день направлены на изучение гипотензивной активности производных изохинолина. Основываясь на влиянии производных изохинолина на процессы свертывания крови, синтезируются новые прекурсоры лекарственных препаратов, в частности тиклид, представляющий собой по структуре тиофеновый аналог N�бензилизохинолина [1]. С целью поиска новых лекарственных средств актуальным остается разработка новых подходов к синтезу гетероциклических соединений ряда изохинолина. Следует отметить также, что на сегодняшний день представляет интерес и изучение особенностей оптической изомерии гетероциклических соединений, а также её влияния на фармакологическую активность лекарственных соединений [2].
Введение углеводных остатков в молекулы подобных гетероциклических биологически активных соединений или природных соединений позволит решить ряд важных научных и практических задач. Так, гликозилирование антибиотиков, противоопухолевых, антивирусных препаратов модифицировало действие исходных гликозил-акцепторов по сравнению с негликозилированными аналогами. Применяемые в этих целях моно- или олигосахариды, как правило, не токсичны для организма человека и животных, что делает такой способ модификации весьма перспективным. Синтезировано большое количество как 1,2-транс-, так и 1,2-цис-гликозидов, пригодных для подобных исследований.
Несмотря на значительные успехи, достигнутые в гликозидном синтезе, универсальных подходов, регио- и стереоселективно, и с высокими выходами приводящих к гликозидам определенного строения, не существует. Поэтому в химии аномерного центра центральное место занимает развитие существующих и разработка новых селективных и эффективных методов построения гликозидной связи [3]. В 60-х годах прошлого века были сформулированы основные принципы межфазного катализа [4-7]. В настоящее время можно сказать, что по широте охвата типов химических реакций катализ в двухфазных системах представляет собой уникальное явление, и широко применяется в тонком и малотоннажном органическом синтезе. Данный подход нашел широкое распространение и в химии углеводов, наряду с известными методами гликозидного синтеза [3-6]. Наряду с очевидным достоинством межфазного катализа – высокие выходы продуктов и стереоспецифичностью, следует отменить и существенные недостатки – использования избытка реагентов и водных растворов оснований, затрудняющих гликозилирование соединений, неустойчивых в этих условиях. Но, несмотря на это, межфазные каталитические процессы остаются весьма ценными в синтетической химии углеводов.
Таким образом, целью настоящей дипломной работы является синтез гликозидов N-ацетил-D-глюкозамина с агликонами пиразолоизохинолинов с использованием каталитической межфазной системы «твердое тело – органический растворитель» для изучения их медико-биологические свойства.
1. МЕТОДЫ СИНТЕЗА ПИРАЗОЛОИЗОХИНОЛИНОВ
Формирование углерод – углеродной связи – один из важнейших процессов в синтетической органической химии. Среди разнообразных методов её образования реакция Пикте–Шпенглера является одной из ключевых реакций в синтезе гетероциклических соединений, которую интенсивно применяют для синтеза, например алкалоидов и их синтетических аналогов на протяжении последних 100 лет. Она основана на кислотно-катализируемой конденсации альдегида или кетона с 2 арил(гетарил)этиламином, ароматический фрагмент которого способен к электрофильной атаке. Последующая циклизация С-нуклеофила (гетеро) ароматического ядра и иминиевого иона приводит к образованию новой углерод – углеродной связи и образование азотсодержащего гетероцикла (схема 1).Однако, несмотря на всю привлекательность этой стратегии, её использование было ограничено только триптамином/триптофаном, гистамином/гистидином, и дофамином/тирамином как аминными субстратами, что неизменно приводит к образованию гетероциклов с базовыми структурами тетрагидро--карболина (ТГБК), тетрогидроимидазопиридина (ТГИ) и тетрагидроизохинолина (ТГИХ) (схема 11) [8, 9]. В настоящее время, несмотря на большое количество работ с использованием реакции Пикте-Шпенглера [10-12], стратегия синтеза осталась неизменной. Даже на твердой фазе, использование которой открыло новые возможности в химии гетероциклов, реакция Пикте-Шпенглера ограничена формированием только классических систем ТГБК, ТГИ и ТГИХ [13-17].
В последние годы эта реакция переживает второе рождение – использование так называемых субстратов Пикте-Шпенглера другого поколения дало возможность значительнее расширить круг получаемых азотсодержащих гетероциклических систем.
В основе нового подхода лежит использование для циклизации не алифатических, а ароматических и гетероциклических аминов – орто-гетарилзамещенных анилинов, орто-арил- и орто-гетарилзамещенных аминогетероциклов.
Схема 1
Можно сказать, что эти исходные соединения по структуре и глубине их превращений не могут быть прямо соотнесены к субстратам реакции Пикте-Шпенглера, и их циклизация на самом деле является новыми кислотно-катализируемыми реакциями. Использование термина «протокол Пикте-Шпенглера», включает в себя одновременно условия, направление циклизации и общее строение получаемых гетероциклов.
Исследование взаимодействия арилзамещенных аминогетероциклов, в том числе 5-амино-4-арилпиразолов, с карбонильными соединениями в условиях модифицированной реакции Пикте-Шпенглера открыло новый путь синтеза конденсированных производных изохинолина. Подавляющее большинство изохинолинов, индоло[2,3-с]-пиридинов и поликонденсированных гетероциклов на их основе получено в результате использования давно открытых и хорошо изученнных реакций – Померанца-Фрича, Бишлера-Напиральского, Пикте-Шпенглера [18].
Авторами работы [18] показаны превращения N-гетарилзамещенных 5�аминопиразолов в условиях реакции диазотирования, а также модифицированной реакции Пикте-Шпенглера, с целью расширения возможностей разработанных ранее методов получения конденсированных азотсодержащих гетероциклов и выяснения влияния гетероциклического заместителя в положении 1 пиразольного цикла на ход реакций и строение конечных продуктов.
Хотя метод получения 5-аминопиразолов из -кетонитрилов и гидразинов является общим и неоднократно описан, условия проведения циклизации часто зависят от свойств применяемых реагентов. Гетарилзамещенные гидразины отличаются меньшей, по сравнению с фенилгидразином, реакционной способностью, а также малой растворимостью в большинстве органических растворителей. Для получения аминопиразолов 12а-f с удовлетворительными выходами потребовалось длительное нагревание реагентов в ледяной уксусной кислоте, поскольку при проведении реакции в течение 1-2 часов выходы продуктов не превышают 20-30% (Табл. 1).Спектральные характеристики аминопиразолов 12а-f соответствуют предложенным структурам. Введение гетарильного заместителя в молекулу 5-аминопиразола приводит к увеличению их температур плавления, по сравнению с N-арилзамещенными [19, 20].
Схема 2
Таблица 1
Время и выходы замещенных N-гетарил-5-аминопиразолов 12а-f
|
№
|
R
|
Het
|
Время, час
|
Выход,%
|
|
12a
|
Me
|
3,5-дихлор-2-пиридил
|
12
|
65
|
|
12b
|
Ph
|
3,5-дихлор-2-пиридил
|
13
|
65
|
|
12c
|
4-ClPh
|
3,5-дихлор-2-пиридил
|
14
|
69
|
|
12d
|
Me
|
бензотиазол-2-ил
|
12
|
70
|
|
12e
|
Ph
|
бензотиазол-2-ил
|
13
|
67
|
|
12f
|
Me
|
4,6-диметилпиримидин-2-ил
|
12
|
60
|
При взаимодействии аминопиразолов 12а-f с нитритом натрия в уксусной кислоте образуются продукты реакции не ионной природы: промежуточно образующиеся соли диазония вступают в реакцию внутримолекулярного азосочетания с образованием соответствующих 3-Неt-1-R-7,8-диметоксипиразоло[3,4-c]циннолинов 14а-f. Выходы соответствующих азоло[3,4-c]циннолинов 14а-f приведены в таблице 2.
Схема 3
Таблица 2
Выходы 3-Неt-1-R-7,8-диметоксипиразоло[3,4-c]циннолинов 14а-f
|
№
|
R
|
Het
|
Выход, %
|
|
14а
|
Me
|
3,5-дихлор-2-пиридил
|
85
|
|
14b
|
Ph
|
3,5-дихлор-2-пиридил
|
41
|
|
14c
|
4-ClPh
|
3,5-дихлор-2-пиридил
|
31
|
|
14d
|
Me
|
1,3-бензтиазол-2-ил
|
45
|
|
14e
|
Ph
|
1,3-бензтиазол-2-ил
|
34
|
|
14f
|
Me
|
4,6-диметилпиримидин-2-ил
|
62
|
В работах [20, 21] авторами было показано, что направление реакции и возможность циклизации определяются как строением карбонильного соединения, так и природой гетероцикла. Поэтому представлялось актуальным изучить реакции N-гетарилзамещенных аминопиразолов с различными карбонильными соединениями, например с 4-хлорбензальдегидом, формальдегидом и изатином. Известно, что алканали, кетоны алифатического, алициклического и ароматического рядов не реагируют с аминопиразолами.
Взаимодействие аминопиразолов 12а, 12d и 12f с 4-хлорбензальдегидом протекает по пути окислительной циклизации, но наличие гетероциклического заместителя в положении 1 пиразольного цикла оказывает влияние на протекание реакции и строение конечных продуктов. Образование пиразоло[3,4-c]изохинолинов 17а, 17b из аминопиразолов 12d и 12f протекает с выходами 45% и 37% соответственно (схема 11). Реакционная масса частично осмоляется, что может быть связано с взаимодействием бензальдегида и аминопиразола по другому пути. Строение полученных 5-арилпиразоло[3,4-c]изохинолинов 17а, 17b подтверждено аналитическими и спектральными методами. Их протонные спектры находятся в соответствии с предполагаемой структурой и данными работы.
Схема 4
При нагревании 5-амино-1-(3,5-дихлорпирид-2-ил)-пиразола (12а) и 4-хлорбензальдегида в трифторуксусной или муравьиной кислоте в течение 1015 часов из реакционной массы были выделены два продукта реакции. Спектральные и аналитические характеристики одного из них соответствуют ожидаемому 1-метил-7,8-диметокси-3-(3,5-дихлорпиридин-2-ил)-5-(4-хлорфенил)пиразоло[3,4-c]изохинолину (19). Данные ИК-спектроскопии и элементного анализа также подтверждают структуру 1-метил-7,8-диметокси-5-(4-хлорфенил)пиразоло[3,4-c]изохинолина (20) (схема 5).
Схема 5
Циклизации аминопиразолов 12а и 12d с параформальдегидом протекают с образованием 5-незамещенных пиразоло[3,4-c]изохинолинов 21 и 22 с невысокими выходами - около 20%.
Схема 6
Продукты взаимодействия аминопиразола 12f с параформальдегидом не выделены. В реакции с изатином продукт циклизации 1-метил-3-(бензотиазол-2-ил)-7,8-диметокси-4,5-дигидропиразоло[3,4-c]изохинолин-5-спиро-3’-(2-оксоиндолина) (53) был получен только с аминопиразолом 12d [18] (схема 7).
Схема 7
В реакции 5-аминопиразолa 12d с параформом впервые получен 5H-пиразоло[3,4-с]изохинолин. Циклизация N-незамещенных аминопиразолов 26a�c сопровождается метилированием атома азота пиразольного цикла. Определено, что алкилирование азота пиразольного кольца происходит перед образованием анелированного пиридинового цикла.
С другими алифатическими альдегидами и кетонами, ацетофенонами аминопиразолы не взаимодействуют и выделяются из реакции неизменными.
Схема 8
С бензальдегидами аминопиразолин реагируют с образованием ароматических 5-арилпиразоло[3,4-с]изохинолинов с выходами от 55 до 80%. Интермедиаты превращения – азометины– могут быть выделены с почти 100% выходом. Классические продукты реакции Пикте-Шпенглера – 4,5�дигидропроизводные – в реакционных смесях не обнаружены, хотя для этого были приняты специальные усилия, в частности защита аминогруппы. Непременным условием циклизации является участие кислорода; в инертной атмосфере азометин остается неизменным.
Схема 9
Окисление азометина кислородом в контролируемых условиях показало, что максимальное поглощение кислорода составляет 2,2 моля на 1 моль азометина.
Конечный результат взаимодействия аминопиразолов с гетероциклическими альдегидами зависит от свойств альдегида и может остановиться на стадиях образования азометина, 5-гетарилпиразоло[3,4 с]изохинолина или, в случае использования индол-3 ил и тиофен-2-альдегида, 5-незамещенного пиразолоизохинолина, который образуется при отщеплении гетероциклического остатка альдегида (схема 10).
Схема 10
Использование протокола Пикте – Шпенглера для циклизации других гетероциклов не всегда дает результат. По этой методологии не получено изоксазоло[5,4-с]изохинолинов, так как в условиях реакции происходит деструкция исходных аминов.
2. ГЛИКОЗИЛИРОВАНИЕ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Первый синтез фенилглюкозида 51в основных условиях осуществлен американским химиком А. Михаэлем в 1879 г. взаимодействием полного ацетата -D-глюкопиранозилхлорида 49с фенолятом натрия 50 в 96% этаноле (схема 11) [5]. Использование фенолят-анионами, в качестве нуклеофильных агентов для полученияО--арилгликозидов сопровождается частичным дезацетилированием как исходных гликозил-доноров, так и целевых продуктов реакции, гидролизом связи С-1 – галоген, что обусловливает низкие выходы получения соответствующих гликозидов. Высокая нуклеофильность фенолят-иона позволяет проводить эту реакцию с высоким выходом и в мягких условиях, причем гидролиз ацилгалогеноз не успевает пройти в заметной степени и не может служить серьезной помехой основной реакции. Конденсация, как правило, приводит к 1,2-транс-гликозидам.
Схема 11
Для синтеза арилгликозидов применялись в основном два метода. Первый основан на сплавлении полных ацетатов сахаров с фенолами в присутствии различных кислотных катализаторов [22]. Второй заключается во взаимодействии фенолятов щелочных металлов с ацилгликозилбромидами или ацилгликозилхлоридами в водном ацетоне или смеси спирт-хлороформ [23]. Известны также многочисленные модификации метода Гельфериха [7].
Ни один из указанных методов не является универсальным. Наиболее общий из них, метод сплавления, применим лишь для низкоплавких монофункциональных фенолов. Второй по значению метод, основанный на использовании фенолятов, не пригоден для синтеза арилгликозидов с электронодонорными заместителями в ядре [23], а также для синтеза производных некоторых сахаров, например рамнозы [24]. Модификации метода Гельфериха не могут быть использованы в случае полифункциональных фенолов [25]. Таким образом, известные методы синтеза страдают серьезными недостатками при гликозилировании полифункциональных фенолов, несущих электронодонорные заместители, а также при распространении их на некоторые классы углеводов, типа пентоз или дезоксигексоз.
Развитие методов межфазного катализа в этой области биоорганической химии позволило получить малодоступные другими методами производные, существенным образом повысить селективность ряда процессов и значительно увеличить выходы продуктов многих химических превращений моносахаридов.
Авторами работы [3] на примере N-ацетилированных 4�нитрофенилгликозидов была показано возможность получения соединений, потенциально являющихся хромогенными субстратами для изучения гиалуронидазы. Гликозилирование проводилось в условиях межфазного катализа в системе «метиленхлорид – 1М раствор карбонат калия» с применением в качестве катализатора тетрабутиламмония гидросульфата (ТБАГ). Замена 1-О-ацильного производного на метилгликозид гиалуроновой кислоты приводило к невысоким выходам 4-нитрофенилглюкозида. Причиной этого является низкая концентрация образующего в реакционной системе -бромида гиалуроновой кислоты.
Схема12
Для качественной и количественной оценки работы ферментов, рассматриваются объекты, как природного происхождения, так и синтетические. Например, агликоноподобные -кетозидазы должны обеспечивать легкое их обнаружение с помощью спектроскопических свойств. Арильные производные N-ацетилнейраминовой кислоты оказались удобными для данных исследований. Ряд методов для стереоселективного получения -кетозидов из N�ацетилнейраминовой кислоты, широко описаны в литературе и содержат ряд недостатков. Например, по методу Кенигса-Kнoрра получение подобных производных требовало применение дорогих катализаторов, предусматривало многостадийный процесс или продолжительный синтез, протекающий зачастую с низкими выходами. Одной из причин невысоких выходов являлась низкая растворимость образующихся солей ароматических соединений в органических растворителях. Проведение реакции в межфазных условиях позволяла исключить эти недостатки. Так, Джордж Ротермель с сотрудниками показал [26], что гликозилирование N�ацетилнейраминовой кислоты в условиях межфазного катализа в системе «хлороформ-водного раствора щелочи» с применением в качестве катализатора бензилтриэтиламмоний хлорида (ТЭБАХ) позволило получить известные и новые арил--кетозиды с высокой стереоселективностью. Реакция протекала быстро и приводила к хорошим выходам арилпроизводных. Например, 4-метилумбеллиферил -кетозид, являющийся стандартным субстратом нейраминидаз, в данных условиях получен с выходом 70%. Для получения 4-метилумбеллифирильного производного необходимо использование 0,1 н. раствора основания. Уменьшение и увеличение концентрации раствора щелочи приводило к протеканию побочных процессов: гидролиза гликозил-галогенида; деструкции 4-метилумбеллиферона. Авторам работы удалось синтезировать новый кетозид флуоресцеин N�ацетилнейраминовой кислоты. Монокетозид получен с выходом 40%, тогда как метод Кенигса-Kнoрра приводил к получению гликозида с выходом ниже 10%. В условиях межфазного катализа идентифицировано образование в небольших количествах побочного продукта – биспроизводного N�ацетилнейраминовой кислоты, выделение которого не представлял затруднений, что еще раз показывает преимущество данного подхода, в отличие от метода Кенигса-Kнoрра.
Схема 13
Взаимодействием ацетобромглюкозы в условиях межфазного катализа получен п�формилглюкозид с выходом 55%. Реакция проводилась в системе хлороформ – водный раствор карбоната калия в присутствии тетрабутиламмоний бромида [26].
Схема14
Авторами работы [4] было показано, что использование р-нитрофенил-2-О--L-фукопиранозил--D-галактопиранозида в качестве субстрата позволяло обнаружить -L-фукозидазу, имеющую специфичность к -(12)-связи. р-Нитрофенил-3,4,6-три-O-ацетил--D-галактопиранозид получен в условиях межфазного катализа. Реакция 3,4,6-три-О-ацетил-2-O-(2,3,4-три-О-ацетил--L-фукопиранозил)--D-галактопиранозилбромида с р-нитрофеноксидом протекала в среде метиленхлорида в присутствии амберлиста А-26 с выходом продукта реакции 34%.
Схема15
В работе [5] авторы показали возможность получение 4-гидроксихинолин--гликозида. Соединения хинина известны как противомалярийные препараты. Анализ синтетических аналогов, обладающих противомалярийным действием, в настоящее время остается актуальным.
Схема16
Замещенные фенилгликозиды широко используется для обнаружения ферментативного расщепления гликозидной связи. В работе [5] был получен ряд замещенных фенил-2-ацетамидо-2-дезокси--и �D�глюкопиранозидов. Замещенные фенил-2-ацетамидо-2-дезокси--D-глюкопиранозиды были получены реакцией -хлорида N�ацетилглюкозамина с фенолятом натрия по методу Кенигса-Кнора. Взаимодействием сполна ацетилированных глюкозамина с различными фенолом в присутствии безводного хлорида цинка получены соответствующие -гликозиды (схема17). Выходы - и -D-глюкопиранозидов приведены в таблице 3.
Схема17
Таблица 3.
Выходы замещенных фенил 2-ацетамидо-2-дезокси-D-глюкопиранозидов
|
Заместители
|
Формула
|
Выход
|
Заместители
|
Формула
|
Выход
|
|
Фенил-3,4,6-три-O-ацетил-2-ацетомидо-2-диокси--D-глюкопиранозиды
|
Фенил-3,4,6-три-O-ацетил-2-ацетомидо-2-диокси--D-глюкопиранозиды
|
|
p-CH3O
|
C21H27O10N
|
17
|
p-CH3O
|
C21H27O10N
|
13
|
|
p-CH3
|
C21H27O9N
|
30
|
p-CH3
|
C21H27O9N
|
27
|
|
m-CH3
|
C21H27O9N
|
24
|
m-CH3
|
C21H27O9N
|
38
|
|
p-Cl
|
C20H24O9NCl
|
17
|
p-Cl
|
C20H24O9NCl
|
8
|
|
m-Cl
|
C20H24O9NCl
|
16
|
m-Cl
|
C20H24O9NCl
|
11
|
Ранее авторами работы синтезирован ряд гликозидов трет-бутил-4-гидрохинона с целью изучения их антиоксидантных свойств. Подобные соединения ингибируют липидное окисление. В качестве исходных гликозил-доноров рассматривались сполна ацетилированные D-глюкоза, D-галактоза и 2-дезокси-2-бутанамидоглюкопиранозилхлорид. Были изучены условия гликозилирования трет-бутил-4-гидрохинона (табл. 4). Во всех случаях реакция протекала с образованием смеси аномеров. Суммарный выход - и -гликозидов составил от 33 до 40%. Образование продуктов с �конфигурации гликозидной связи наблюдалось в большей степени (табл. 4). В качестве побочного продукта идентифицирован гликозид 72 с выходом 2% (схема 18). Использование трифлата иттербия (Yb(OTf)3) уменьшало время реакции, но не влияет на выход продуктов и стереоселективность реакции. Взаимодействием пентаацетата -D-галактозы 70b с замещенным гидрохиноном получен соответствующий -галактозид с выходом 40% (табл. 4). Введение 2-дезоксисахара в молекулу двухатомного фенола стало возможным при использовании -D-глюкопиранозилхлорида. 2-Аминогликозид гидрохинона получен с выходом 60%.
Схема18
Таблица 4.
Выходы гликозидов трет-бутилгидрохинона
|
Донор
|
Условия
|
Продукт
|
Суммарный выход,(%)
|
Соотношение /
|
|
69a
|
(p-СH3)PhSO3H,125оС
|
71a/71a+72
|
33+2
|
29/71
|
|
69a
|
(p-СH3)PhSO3H, CH2Cl2
|
71a/71a
|
67
|
4/96
|
|
69a
|
(p-СH3)PhSO3H, Yb(OTf)3, CH2Cl2
|
71a
|
61
|
10/90
|
|
70b
|
(p-СH3)PhSO3H, CH2Cl2
|
71b/71b
|
40
|
15/85
|
|
70c
|
ZnCl2, CH2Cl2
|
71c/71c
|
60
|
11/89
|
|
70b
|
ZnCl2, CH2Cl2
|
71d/71d
|
40
|
5/95
|
3. МАТЕРИАЛЫ И МЕТОДИКА ИССЛЕДОВАНИЯ
Температуры плавления определяли на приборе ПТП, оптическое вращение при 20-25°С – на поляриметре Polamat-A ( = 546 нм).
Анализ состава реакционных смесей, чистоты синтезированных соединений, а также мониторинг реакций, проводили методом тонкослойной хроматографии (ТСХ) на пластинках Sorbfil-АФВ-УФ («Сорбполимер», Россия). Для проявления хроматограмм использовали системы растворителей: бензол–изопропиловый спирт, 10:1(А), хлороформ – изопропиловый спирт 15:1 (B). Зоны веществ обнаруживали 5% раствором серной кислоты в этаноле при нагревании до 200-300 оС или УФ-облучением при длине волны 254 нм. Выделение индивидуальных веществ осуществляли колоночной хроматографией на силикагеле Kieselgel 60 (0.063-0.200 мм).
1Н ЯМР спектры получены на спектрометрах Varian Mercury-400 (400 МГц), внутренний стандарт – Me4Si. Приведены химические сдвиги (ХС) (м.д., -шкала) и константы спин-спинового взаимодействия (КССВ, J, Гц).
Использовали 15-краун-5 (Merck, Германия), карбонат калия (хч), пропан-2-ол (ч) (Технохим), бромид ртути (Технохим), ацетонитрил (Сфера Сим, чда), хлороформ (чда), ацетон (чда), бензол (чда) (Крымлаборреактив).
Пиразо[4,5-c]изохинолины 74, 76, 78, 80 синтезированы отделом химии биологически активных соединений Институтом физико-органической химии и углехимии им. Л.М. Литвиненко НАН Украины.
Синтез 8-[4-(2-aцетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкопиранозилокси)фенил]-3-метил-5,6-диметокси-1-фенилпиразо[4,5�c]изохинолина (75)
Схема 19
Смесь 500 мг (1,4 ммоль) -хлорида 73, 562 мг (1,4 ммоль) пиразолоизохинолина 74, 869 мг (6,3 ммоль) безводного карбоната калия и 54 мкл (0,28 ммоль) 15-краун-5 в 15 мл безводного ацетонитрила перемешивали при температуре 20-22 С до полной конверсии гликозил-донора (ТСХ, система A). Твердую фазу отделяли фильтрованием, осадок промывали на фильтре ацетонитрилом (25 мл), растворитель удаляли досуха при пониженном давлении. Выход продукта 75 после колоночной хроматографии (градиентное элюирование: хлороформ–изопропиловый спирт, 100:1 хлороформ–изопропиловый спирт, 30:1) составил 700 мг (66%); т.пл. 274 оС, []546 -25о (c 1,0; хлороформ).
1Н ЯМР (DMSO-d6, 400 МГц): 1,83с (3H, NAc), 1,98с (3H, OAc), 2,03с (6H, OAc), 3,79с (3Н, ОCH3), 4,10м (5Н, Н-2, Н-6аb, ОCH3), 4,23м (2Н, Н-5, Н-6аb), 4,97дд (1Н, Н-4, J4,5 10,0 Гц), 5,27дд (1Н, Н-3, J3,4 10,0 Гц), 5,52д (1Н, Н-1, J1,2 8,0 Гц), 7,28дд (3Н, NPh),. 7,51с (1Н, СНаром.), 7,54д (2Н, NPh), 7,72с (1Н, СНаром.), 7,8д (2Н, СНаром.), 8,16д (1Н, NН, J2,NH 8,0 Гц), 8,32д (2Н, СНаром.).
Синтез 8-[4-(2-aцетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкопиранозилокси-3-метокси)фенил]-5,6-диметокси-1-фенил-3-этилпиразо[4,5-c]изохинолина (77)
Схема 20
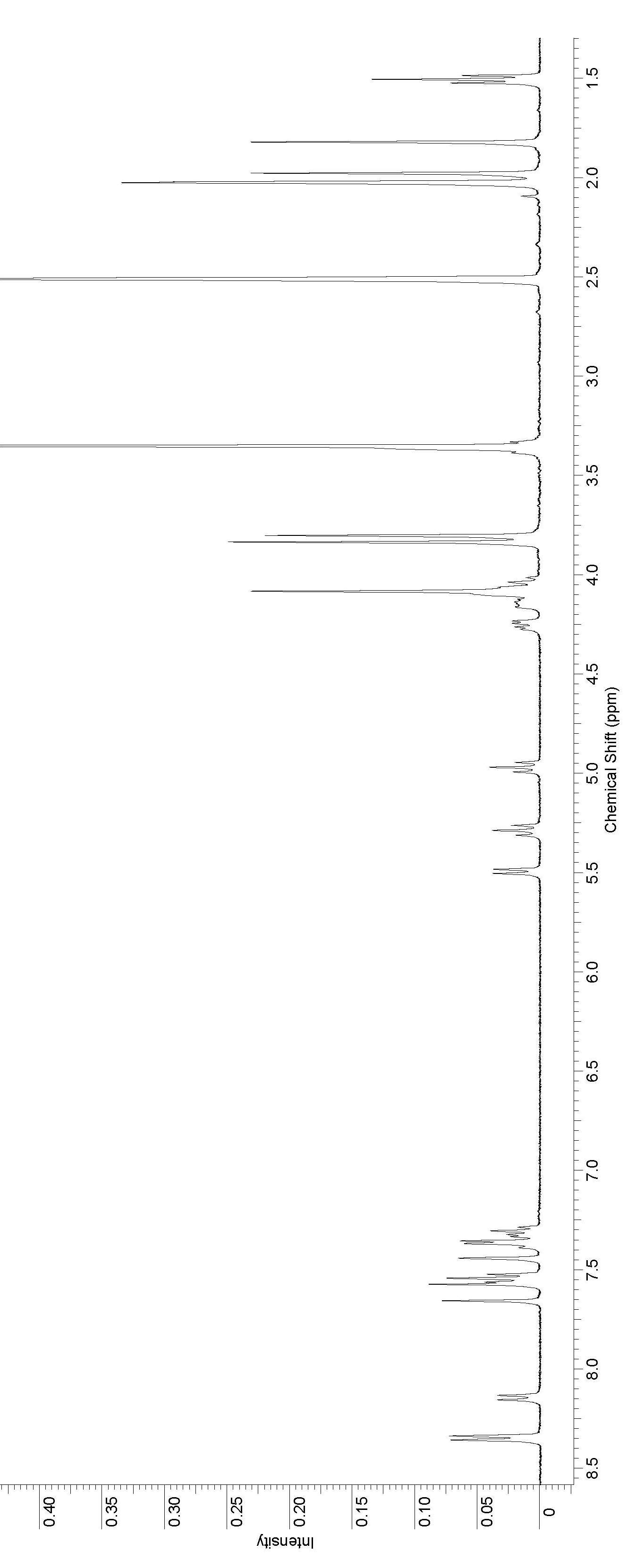
Аналогично, по методике синтеза соединения (75) из 400 мг (1,1 ммоль) -хлорида 73 и 483 мг (1,1 ммоль) пиразолоизохинолина 76 получили продукт 77 с выходом 420 мг (49%); т.пл. 230оС, []546 -6,25о (c 1,0; хлороформ).
1Н ЯМР (DMSO-d6, 400 МГц): 1,50т (3Н, СН3), 1,82с (3H, NAc), 1,98с (3H, OAc), 2,03с (6H, OAc), 3,80с (3Н, ОCH3), 3,83с (3Н, ОCH3), 4,08м (6Н, H-2, Н-5, Н-6аb, ОCH3), 4,25дд (1Н, Н-6аb, J6a,6b 12 Гц), 4,97дд (1Н, Н-4, J4,5 10,0 Гц), 5,29дд (1Н, Н-3, J3,4 10,0 Гц), 5,49д (1Н, Н-1, J1,2 8,0 Гц), 7,35м (3Н, NPh),. 7,44с (1Н, СНаром.), 7,54м (3Н, NPh, СНаром.), 7,66с (1Н, СНаром.), 8,15д (1Н, NН, J2,NH 12,0 Гц), 8,35д (2Н, СНаром.).
Синтез 8-[4-(2-aцетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкопиранозилокси)фенил]-5,6-диметокси-1-фенил-3-этилпиразо[4,5�c]изохинолина (79)
Схема 21
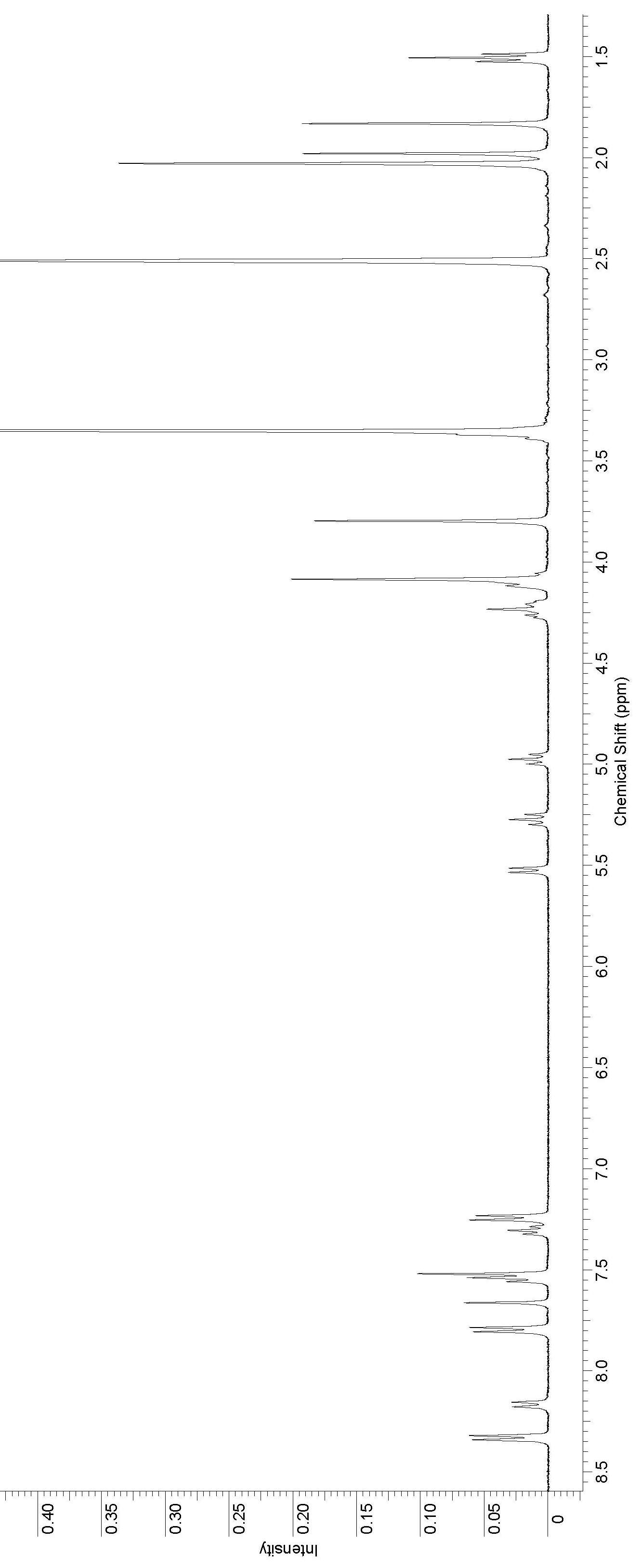
Аналогично, по методике синтеза соединения (75) из 500 мг (1,4 ммоль) -хлорида 73 и 562 мг (1,4 ммоль) пиразолоизохинолина 78 получили продукт 79 с выходом 636 мг (60%); т.пл. 269 оС, []546 -8,33о (c 1,0; хлороформ).
1Н ЯМР (DMSO-d6, 400 МГц): 1,51т (3Н, СН3), 1,83с (3H, NAc), 1,98с (3H, OAc), 2,03с (6H, OAc), 3,8с (3Н, ОCH3), 4,08м (5Н, Н-2, H-6ab, ОCH3), 4,23м (2Н, Н-5, H-6ab), 4,97дд (1Н, Н-4, J4,5 10,0 Гц), 5,27дд (1Н, Н-3, J3,4 10,0 Гц), 5,55д (1Н, Н-1, J1,2 8,0 Гц), 7,27м (3Н, NPh),. 7,52с (1Н, СНаром.), 7,55д (2Н, NPh.), 7,66с (1Н, СНаром.), 7,8д (2Н, СНаром.), 8,17д (1Н, NН, J2,NH 12,0 Гц), 8,33д (2Н, СНаром.).
Синтез 8-[4-(2-aцетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкопиранозилокси)фенил]-3-метил-5,6-диметокси-пиразо[4,5�c]изохинолина (81)
Схема 22
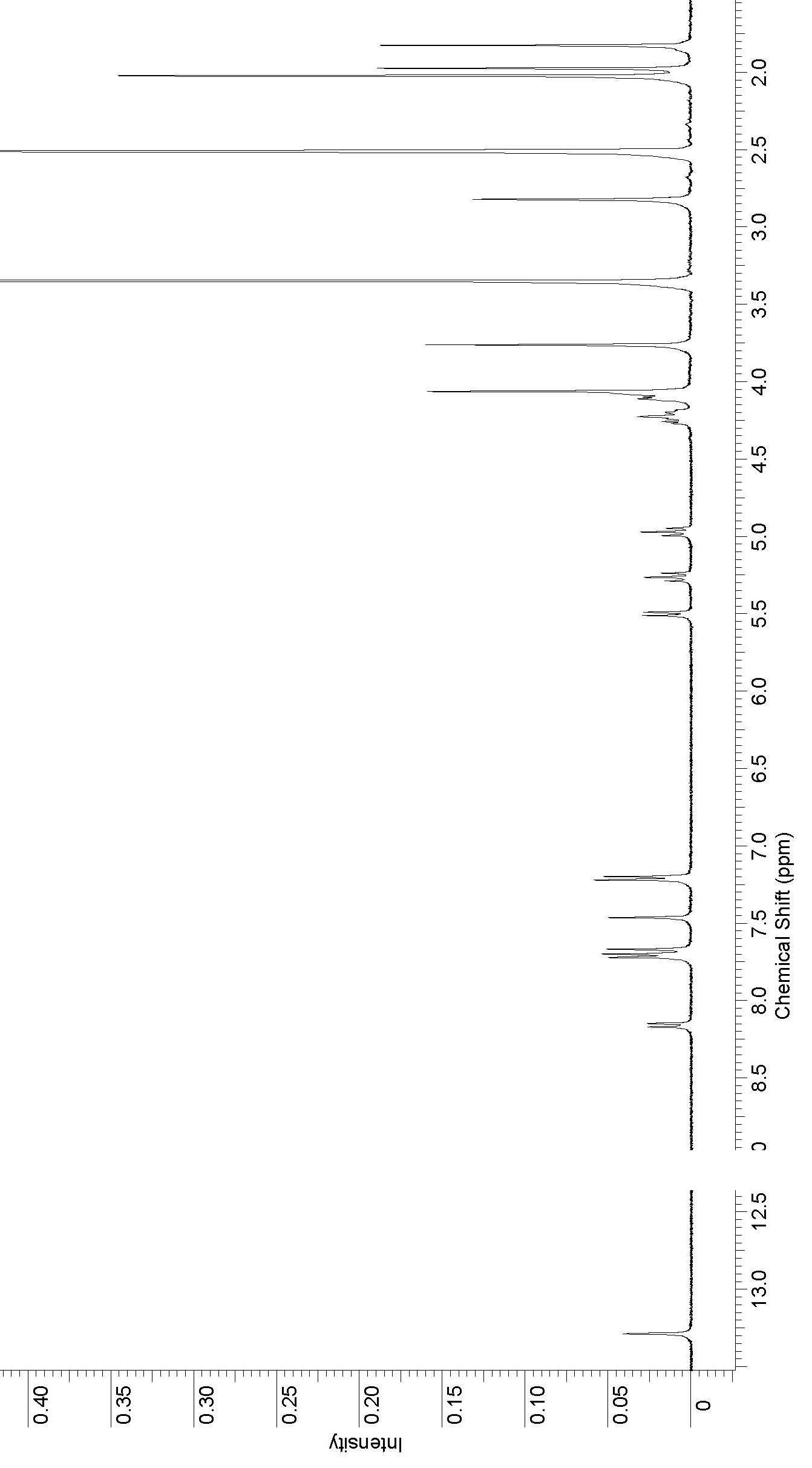
Аналогично, по методике синтеза соединения (75) из 300 мг (0,8 ммоль) -хлорида 73 и 288 мг (0,8 ммоль) пиразолоизохинолина 80 получили продукт 81 с выходом 230 мг (42%); т.пл. 248 оС, []546 -8,33о (c 1,0; хлороформ).
1Н ЯМР (DMSO-d6, 400 МГц): 1,83с (3H, NAc), 1,97с (3H, OAc), 2,02с (6H, OAc), 2,82 с (3Н, СН3), 3,76с (3Н, ОCH3), 4,08м (5Н, Н-2, H-6ab, ОCH3), 4,23м (2Н, Н-5, H-6ab), 4,97дд (1Н, Н-4, J4,5 8,0 Гц), 5,26дд (1Н, Н-3, J3,4 10,0 Гц), 5,50д (1Н, Н-1, J1,2 8,0 Гц),. 7,21д (2Н, СНаром.), 7,46с (1Н, СНаром.), 7,67с (1Н, СНаром.), 7,71д (2Н, СНаром.), 8,16д (1Н, NН, J2,NH 8,0 Гц), 13,29с (1Н, NH).
Синтез 1-(2-aцетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкопиранозил)-3,8-диметил-5,6-диметокси-пиразо[4,5�c]изохинолина (83)
Схема 23
В 10 мл безводного толуола растворили 450 мг (1,2 ммоль) -хлорида 73 и 300 мг (1,2 ммоль) пиразолоизохинолина 82. В реакционную смесь внесли 471 мг (1,3 ммоль) бромида ртути (II) и кипятили до полной конверсии -хлорида 73. (ТСХ, система A). Твердую фазу отделяли фильтрованием, осадок промывали на фильтре толуолом (25 мл), растворитель удаляли досуха при пониженном давлении. Выход продукта 83 после колоночной хроматографии (градиентное элюирование: хлороформ–изопропиловый спирт, 100:1 хлороформ–изопропиловый спирт, 30:1) составил 72 мг (10%); т.пл. 270-275 оС, []546 -25о (c 1,0; хлороформ).
1Н ЯМР (DMSO-d6, 400 МГц): 1,48с (3H, NAc), 1,97с (3H, OAc), 1,98c (3H, OAc), 2,03с (3H, OAc), 2,76c (3Н, СН3), 2,93с (3Н, СН3), 3,96м (5Н, Н-2, H-6ab, ОCH3), 4,03с (3Н, ОСН3), 4,21м (2Н, Н-5, H-6ab, J6a,6b 12,0 Гц), 5,04дд (1Н, Н-4, J4,5 10,0 Гц), 5,53дд (1Н, Н-3, J3,4 10,0 Гц), 6,49д (1Н, Н-1, J1,2 8,0 Гц), 7,55с (1Н, СНаром.), 7,56с (1Н, СНаром.), 8,00д (1Н, NН, J2,NH 8,0 Гц).
4. ОХРАНА ТРУДА И БЕЗОПАСНОСТЬ В ЧЕРЕЗВЫЧАЙНЫХ
СИТУАЦИЯХ
Дипломная работа выполнялась в лаборатории кафедры органической и биологической химии Таврического национального университета имени В.И. Вернадского, оборудованной с соблюдением всех правил охраны труда. При работе неукоснительно соблюдались правила техники безопасности, обязательные для химической лаборатории.
При выполнении дипломной работы нами использовались следующие вещества: 2-ацетамидо-3,4,6-три-О-ацетил-2-дезокси--D-глюкозаминил-хлорид, изихинолин (C9H7N), карбонат калия (K2CO3), хлороформ (CHCl3) ацетонитрил (CH3CN), пропан-2-ол (CH3-CH(OH)-CH3),ацетон (CH3-C(O)-CH3), бромид ртути (HgBr2), краун-эфир (15К5) [27-30].
Общие правила техники безопасности:
1) В химической лаборатории необходимо содержать рабочее место в чистоте и порядке, следить за исправностью аппаратуры и приборов. Перед уходом из лаборатории убрать рабочее место,
2) Перед началом работы необходимо надеть спецодежду и иметь индивидуальные средства защиты, предусмотренные инструкцией.
3) Каждый работающий должен знать, где в лаборатории находятся противопожарные средства и аптечка, содержащая всё необходимое для оказания первой медицинской помощи.
4) Необходимо соблюдать меры предосторожности, указанные в специальной инструкции по технике безопасности и в методическом руководстве.
5) В лаборатории категорически запрещается принимать пищу, хранить продукты, пить, курить. Перерывы работы, связанные с приёмом пищи и курением, делаются с ведома руководителя и только после тщательного мытья рук.
6) Категорически запрещается работать в лаборатории одному.
7) Все опыты с ядовитыми веществами проводятся в вытяжном шкафу.
8) Категорически запрещается держать в лаборатории вещества и растворы в посуде без надписи названия вещества и концентрации.
9) Требования безопасности по окончанию работы:
• Отключит от сети все приборы, устройства.
• Убрать с рабочих мест реактивы, растворы.
• Тщательно вымыть использованную посуду, колбы, пробирки.
• Произвести влажную уборку помещений.
• Проветрить помещение.
Техника безопасности при работе с кислотами и щелочами:
1) Склянки с кислотами и щелочами необходимо переносить только в специальных ящиках, выложенных асбестом.
2) Работающим с кислотами и щелочами необходимо пользоваться резиновыми перчатками и предохранительными очками.
3) Переливать кислоты и щелочи в мелкую тару необходимо с помощью сифона или ручного насоса.
4) Помнить о том, что при приготовлении растворов серной кислоты ее необходимо переливать в воду тонкой струйкой при непрерывном перемешивании.
5) Растворять щелочи необходимо путем медленного прибавления к воде небольших кусочков, при непрерывном перемешивании, куски щелочи необходимо брать только щипцами.
Техника безопасности при работе со стеклом:
1) Тонкостенную посуду нельзя нагревать на открытом огне, следует использовать асбестовую сетку.
2) Большие химические стаканы с жидкостью следует поднимать только двумя руками, поддерживая одной рукой дно.
3) При разламывании надрезанных стеклянных трубок их нужно растягивать, а не сгибать.
4) Вставляя стеклянную трубку в резиновую пробку, нужно ее ввинчивать, смочив водой, вазелином или глицерином. Конец трубки должен быть оплавлен.
4.1. Карбонат калия
При работе с карбонатом калия наблюдается изъявления слизистой носа. Вдыхание пыли может вызвать раздражение дыхательных путей, конъюнктивит. Происходит снижение функции дыхательного аппарата.
ПДК 2 мг/м3 (по степени воздействия на организм человека относится к веществам 3-го класса опасности).
Меры безопасности: не допускать попадания на кожу. Все работы проводить в резиновых перчатках.
Способы защиты: производственные помещения и лаборатории, в которых проводится работа с углекислым калием, должны быть оборудованы приточно-вытяжной механической вентиляцией.
4.2. Ацетонитрил
Не зависимо от путей поступления в организм при остром отравлении наблюдается вначале головная боль, апатия, тошнота, головокружение, бледность, падение температуры и кровяного давления, судороги, потеря сознания. После выздоровления в течение некоторого времени (до 3 недель) — депрессия, головная боль, сердцебиение, особая слабость мышц верхних конечностей, повышенный диурез, белок в моче, повышенное содержание цианидов в крови и роданидов в моче.
Ацетонитрил менее токсичен, чем ряд других нитрилов жирного ряда. Как полагают, действие ацетонитрила определяется целой молекулой в комбинации с медленно отщепляющейся СN-группой. Характерны вызываемые ацетонитрилом, судороги, влияние на щитовидную железу и вызываемый им отек легких.
В организме ацетонитрил предположительно превращается в роданиды, муравьиную кислоту и аммиак. Возможно окислительное разрушение.
ПДК 10 мг/м; ЛД50 1670 мг/кг (по степени воздействия на организм человека относится к веществам 2-го класса опасности).
Меры предосторожности: избегать попадания на кожу.
4.3. Хлороформ
Наркотик, действующий токсически на обмен веществ и внутренние органы, в особенности на печень. Смерть при отравлении хлороформом обычно наступает от прекращения дыхания. При очень высоком содержании хлороформа в воздухе возможна смерть от остановки сердца.
Острое отравление может вызвать тяжелые последствия и смерть через некоторое время после вдыхания. В более легких случаях наблюдаются рвота, головокружение, слабость, желудочные боли, возбужденное состояние. В крови — анемия, лейкоцитоз.
Даже в относительно невысоких концентрациях хлороформ может вызвать профессиональное хроническое отравление с преимущественным поражением печени. При частом попадании на кожу может вызывать дерматиты, экземы.
Наркотическая концентрация, вызывающая изменение скорости развития рефлекторного мышечного напряжения: 0,25—0,5 мг/л при 40-минутиом вдыхании.
ПДК 120 мг/м3, класс опасности - 2.
Меры предосторожности: избегать попадания на кожу. Все работы проводить под тягой.
Способы защиты: резиновые перчатки, нарукавники, фартуки, противогаз.
4.4. Изопропиловый спирт
Действуют сходно с этиловым спиртом, но при равной концентрации паров сильнее его. Пары изопропилового спирта раздражают слизистые глаз и верхних дыхательных путей.
Работающие с изопропиловым спиртом жалуются на резь в глазах слезотечение, светобоязнь, обостряющиеся со временами. Отмечались сужение поля зрения и конъюнктивит; признаки неврита зрительного нерва с понижением остроты зрения. Восстановление наступило после 2 недель лечения и прекращения работы.
ПДК 10 мг/м3, класс опасности - 2.
Меры предосторожности: фильтрующий противогаз, местная и общая вентиляция.
4.5. Ацетон
Наркотик, последовательно поражающий все отделы центральной нервной системы. При вдыхании в течение длительного времени накапливается в организме; токсический эффект зависит не только от концентрации, но и от времени действия. Медленное выделение из организма увеличивает возможность хронического отравления.
Ацетон может попадать в организм в виде паров при дыхании, а может всасываться через кожу. На участках кожи, подвергавшихся в течение рабочего дня воздействию ацетона, уменьшается рН и количество холестерина, угнетается функция сальных желез, что приводит к покраснению.
ПДК 200 мг/м3, класс опасности - 3.
Меры предосторожности: избегать попадания на кожу. Все работы проводить под тягой.
4.6. Бромид ртути (II)
Интоксикация солями ртути проявляется головной болью, саливацией, покраснением, набуханием и кровоточивостью десен, появлением на них темной каймы сульфида ртути, стоматитом, набуханием лимфатических и слюнных желез. Часто повышается температура. Возникают расстройства пищеварения. В сравнительно легких случаях через 2-3 недели нарушенные функции восстанавливаются, в тяжелых – развиваются резкие изменения в почках (некротический нефроз). В последнем случае через 5-6 дней наступает смерть.
ПДК 0,1 мг/м3, класс опасности - 1.
Меры предосторожности: избегать попадания на кожу. Все работы проводить под тягой.
4.7.Краун-эфиры
Предварительные результаты по испытанию токсичности на мышах по-казывают, что при разовом приеме внутрь доза 300 мг/кг для жизни не опасна [31].
4.8. Электробезопасность
При выполнении работы использовалось следующее электрооборудование: лабораторные электроплитки, магнитные мешалки ММ-5, компьютер. Приборы были снабжены изоляцией и заземлением, необходимым для безопасного обращения.
Воздействия электрического тока на организм могут быть следующими:
1. Тепловое действие. Следствием теплового действия электрической дуги, раскаленной спирали нагревательного прибора и т.д. являются ожоги кожного по-крова, практически не отличающиеся от термических ожогов.
2. Химическое действие. Прохождение тока через электролит – плазму крови – ведет к изменению её состава и разрушению эритроцитов. Результатом этого вида воздействия является нарушение обмена веществ.
3. Биологическое действие проявляется обычно ярче, чем другие виды, и ве-дет к нарушению режима дыхания, частоты сердечных сокращений. Примерно через 0,5-2 с от начала контакта может наступить фибрилляция и кровообращение практически останавливается. Работа сердца прекращается в результате повреждения нервных клеток, управляющих его иннервацией.
Порядка 70-75 % всех электротравм приходится на электроудар, связанный с биологическим воздействием тока на организм. Предельно допустимым током считается ток 0,005 А. Установлено, что большая часть электротравм происходит от напряжения переменного тока в 220 В при непосредственном контакте с оголенными проводами и в условиях повышенной влажности через мокрую одежду, по причине высокой её токопроводимости.
Меры предосторожности: не допускать работу с неисправными, незаизолированными электроприборами. Не допускать попадания влаги внутрь электроприборов. Категорически запрещается прикасаться к оголенным токоведущим жилам и нагревательным элементам, когда прибор включен в электросеть
4.9. Техника безопасности при работе с персональным компьютером
1) Требования безопасности перед началом работы: [32]:
• Подготовить рабочее место, убедиться в достаточной освещенности.
• Убедиться в исправности компьютера, произведя его внешний осмотр. При осмотре обращать внимание на наличие и исправность предусмотренных защитных устройств токоведущих частей, исправность коммутационных устройств кнопок, клавиш, целостность изоляции питающего кабеля, вилок, розеток.
2) Требования безопасности во время работы:
• Выполнять при работе требования, изложенные в руководстве по эксплуатации компьютера.
• Не оставлять компьютер включенным при уходе с рабочего места. При длительном перерыве в работе компьютер следует обесточить, отсоединив от сети.
• При появлении неисправностей прекратить работу, компьютер отключить от электросети (или поступить в соответствии с требованиями руководства по эксплуатации).
• При работе с текстами на бумаге, листы надо располагать как можно ближе к экрану, чтобы избежать частых движений головой и глазами при переводе взгляда. Подставку с документами необходимо установить в одной плоскости с экраном и на одной с ним высоте.
• Во время регламентированных перерывов с целью снижения нервно-эмоционального напряжения, зрительного и общего утомления целесообразно выполнять комплексы упражнений, рекомендованных санитарными нормами и правилами.
Компьютер необходимо установить на столе, где достаточно места не только для монитора и клавиатуры, но и также – для размещения документов. При этом расстояние от края стола до клавиатуры не может быть менее 30 см, а от экрана монитора до работника – менее 50 сантиметров. Сам монитор должен находиться на уровне глаз.
Необходимо помнить – постоянную работу за компьютером ни в коем случае нельзя исполнять без регламентированных перерывов. Причем при 8-часовой смене перерывы на 15-20 минут должны делаться через каждые два часа работы. А в последние четыре часа 12-часовой смены – через каждые 60 минут.
5. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Ранее сотрудниками кафедры органической и биологической химии было показано успешное применение межфазной системы «твердый К2СО3 – безводный CH3CN» с использованием краун-эфира для получения широкого ряда -D-глюкозаминидов с агликонами различной природы [33-36]. Этот подход позволил синтезировать фенилглюкозаминиды, несущие в о-, м- и п-положениях ароматического ядра различные гетероароматические радикалы, такие как 1,3,4-оксдиазол-5-ил, хинолин-4-ил, хиназолин-4-ил. Таким образом, обсуждаемый способ построения 1,2-транс-глюкозаминидной связи может быть эффективным инструментом для введения углеводных остатков в молекулы гетероциклических соединений на основе изохинолина – пиразолоизохинолинов, и позволит получить глюкозаминиды для дальнейшего изучения спектра их биологических свойств.
Глюкозаминилирование пиразолоизохинолинов 74, 76, 78, 80 -D-глюкозаминилхлоридом 73 проводили в межфазной системе «твердый К2СО3 – безводный CH3CN» с использованием катализатора 15К5 при комнатной температуре. Реакция протекала в течение 2-3 ч при стехиометрическом соотношении гликозил-донора и пиразолоизохинолина, 4,5-кратном избытке основания (по субстрату – хлориду 73) и 20 моль% краун-эфира. Во всех случаях, в реакционной среде по данным тонкослойной хроматографии присутствовали следовые количества оксазолина и ряд не идентифицированных продуктов деструкции углеводов. Выходы глюкозаминидов 75, 77, 79, 81 после колоночной хроматографии составили 49-66%.
Строение целевых соединений 75, 77, 79 доказано 1Н ЯМР � спектроскопией. В 1Н ЯМР спектрах соединений 75, 77, 79 однозначно идентифицированных как О--глюкозаминиды (дублеты аномерных протонов в области 5,49-5,55 м.д. с КССВ 8,0 Гц). В спектрах также идентифицированы сигналы скелетных протонов, протонов О- и N-ацетильных защитных групп углеводного остатка, сделано отнесение сигналов как протонов углеводного остатка, так и протонов агликонов.
Пиразолоизохинолин 80, содержащий свободный атом азота пиразольного цикла, в реакции гликозилирования может привести к образованию второго основного продукта реакции - бис-производного с различной природой гликозидной связи. Было обнаружено, что конверсия -D-глюкозаминилхлорида 73 в условиях межфазного катализа сопровождалась образованием только одного основного продукта О-гликозида 81, выход которого составил 42%. В данных условиях атом азота пиразольного цикла не участвовал в реакции, что однозначно подтверждено наличием в1Н ЯМР спектре глюкозаминида 81 синглета протона группы NH пиразольного цикла с ХС 13,29 м.д. 1,2-транс-Диаксиальное расположение протонов в остатке N-ацетилглюкозамина подтверждается величиной КССВ 8,0 Гц и ХС 5,50 м.д. Сравнение области протонов углеводного остатка в 1Н ЯМР спектре гликозида 75 с аналогичными областями в спектрах соединений 75, 77, 79 показало однотипное расположение сигналов.
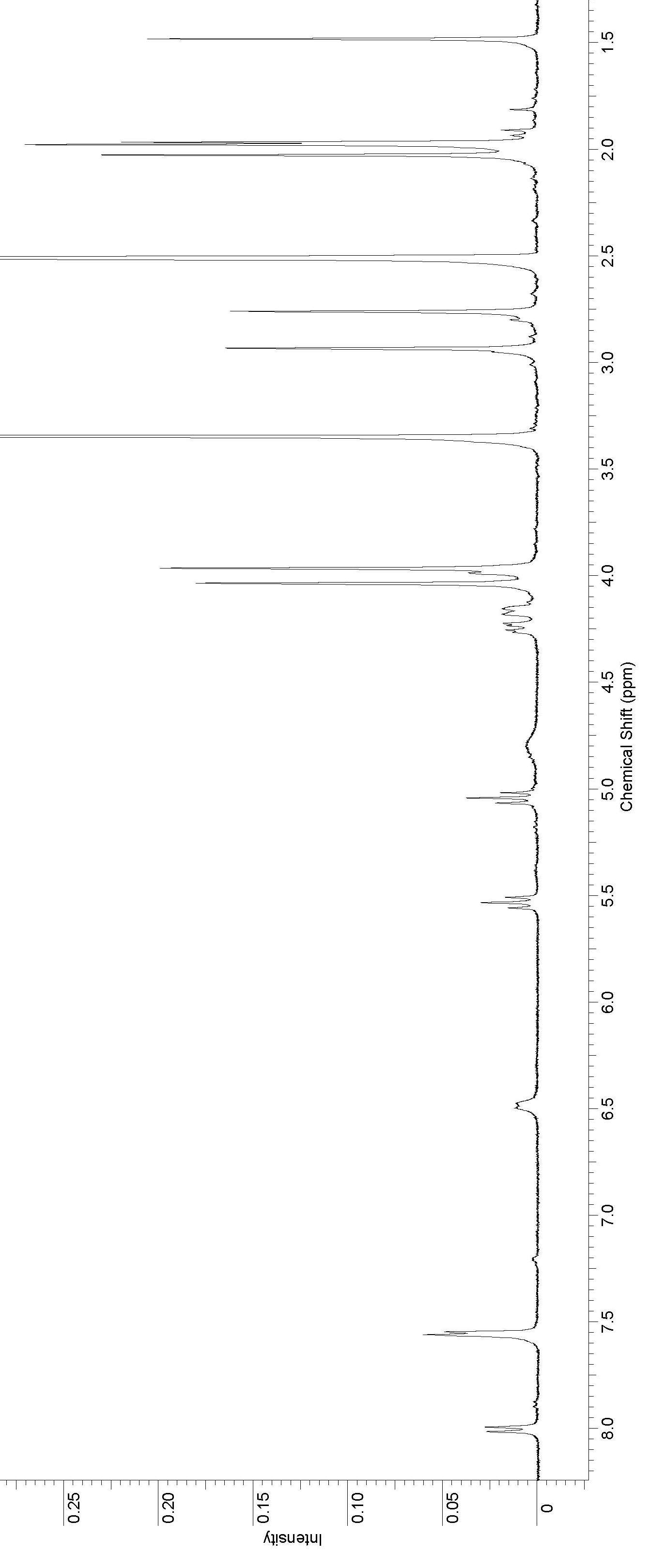
Для введения остатка N-ацетилглюкозамина по свободному атому азота пиразольного цикла, реакцию -D-глюкозаминилхлорида 73 с пиразолоизохинолином 82 проводили с использованием бромида ртути(II) [37, 38]. Выход целевого N-гликозида 83 оказался невысоким – 10%, что связано с заметной деструкцией гликозил-донора в условиях реакции. Таким образом, очевидна перспективность продолжения исследований в данном направлении для оптимизации условий реакции глюкозаминилирования пиразолоизохинолина 82. В 1Н ЯМР спектре N--гликозида 83 наблюдалось значительное смещение в слабое поле сигнала аномерного протона ( 6,49 м.д.) по сравнению с дублетами Н-1 глюкозаминидов пиразолоизохинолинов 75, 77, 79, 81 (рис. 1). При этом КССВ составила 8,0 Гц. В спектре идентифицированы сигналы скелетных протонов, протонов О- и N-ацетильных защитных групп углеводного остатка, а также сигналов ароматических протонов агликона с ХС 7,55 и 7,56 м.д.
Таким образом, пиразолоизохинолины 74,76,78, 80 являются удобными объектами исследования межфазных процессов гликозилирования с целью получения глюкозаминидов для изучения их медико-биологических свойств.
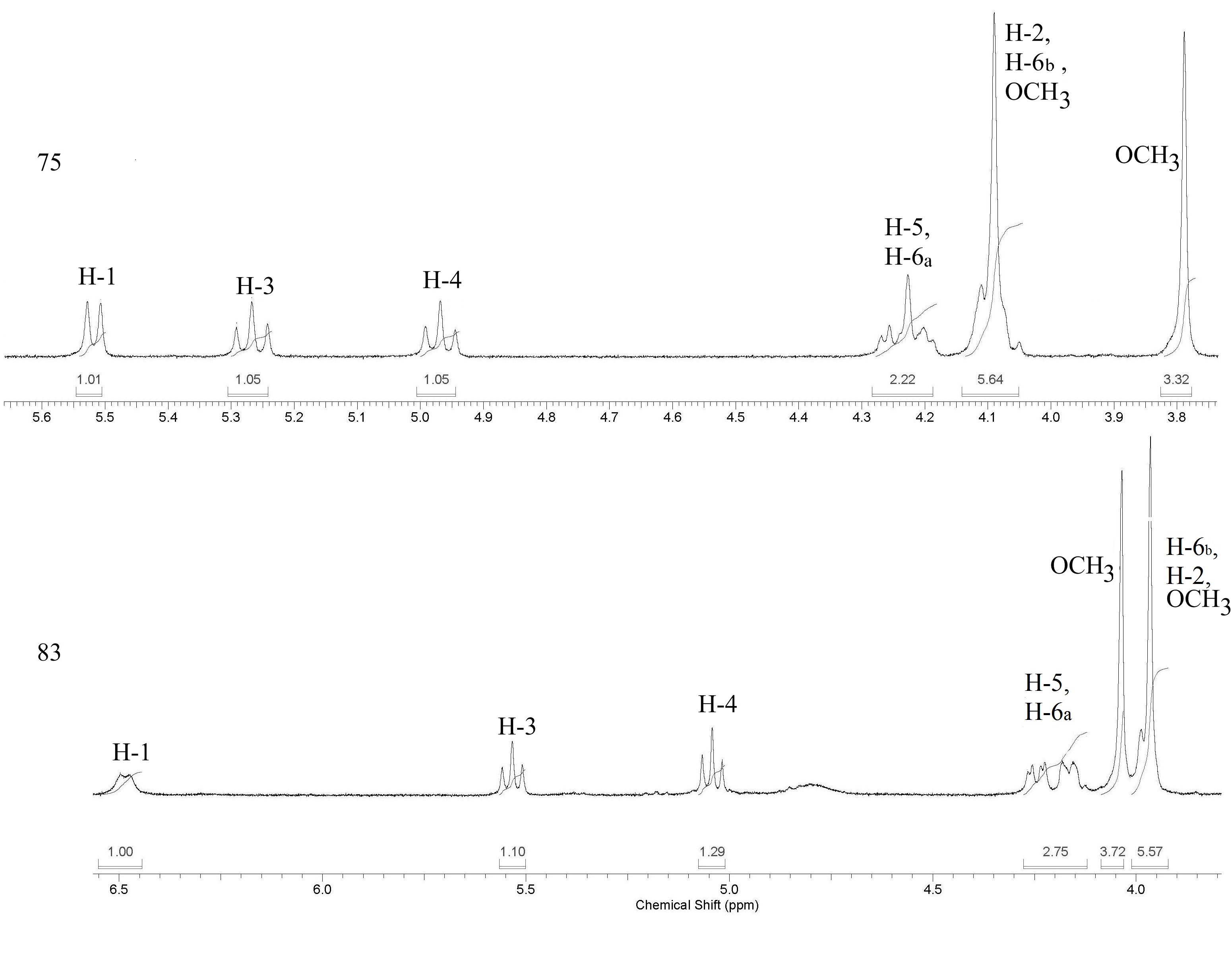
Рис. 1. Область сигналов скелетных протонов в 1Н ЯМР спектрах соединений 75 и 83.
ВЫВОДЫ
- Осуществлен синтез в условиях межфазного катализа глюкозаминидов пиразолоизохинолинов.
- Установлено, что в обсуждаемом межфазном процессе наблюдается образование только O--D-2-ацетамидо-2-дезоксиглюкопиранозидов пиразолоизохинолинов.
- Получен новый N--глюкозаминид пиразолоизохинолина.
СПИСОК ЛИТЕРАТУРЫ
1. Использование производных изохинолина в качестве лекарственных средств
http://www.pharmasvit.com/v3/Spravochniki/2134.html
2. Алексеев В. В. Оптическая изомерия и фармакологическая активность лекарственных препаратов / В. В. Алексеев // Военно-медицинская академия. Журн. – 1998.– № 1. – С. 49-55.
3. Phase-transfer catalyzed synthesis of acetylated aryl -D-glucopyranosides and aryl -D-galactopyranosides / D. Dess, H. Kleine, D. Weinderg [et al.] // Synthesis. – 1981. – № 11. – P. 883-885.
4. Roy R. Stereospecific synthesis of aril -D-N-acetylglucopyranosides by phase-transfer catalysis / R. Roy, F. Tropper // Synth. Commun. – 1990. – Vol. 20, № 14. – P. 2097-2102.
5. Lewis P. T. Regiospecific 4-O--glucosidation of isoflavones / P. T. Lewis, K. Whl // Tetrahedron Lett. – 1998. – Vol. 39. – P. 9559-9562.
6. Грагеров И. П. Краун-соединения в органическом синтезе./ И. П. Грагеров. – Киев: Наукова думка, 1994. – 345 с.
7. Jensen K. J. O-Glycosylations under neutral or basic conditions / K. J. Jensen // Chem. Soc., Perkin Trans. – 2002. – № 1. – P. 2219-2233
8. Royer J. Chiral heterocycles by iminium ion cyclization / J. M. Royer, L. Bonin // – Chem. Rev. – 2004. – Vol. 104, № 5. – P. 2311-2352.
9. Cox E. D. The Pictet-Spengler condensation: a new direction for an old reaction / E. D. Cox, J. Cook // – Chem. Rev. – 1995. – Vol. 95, № 9. – P. 1797-1842.
10. Tsuji R. An efficient synthetic approach to optically active -carboline derivatives via Pictet–Spengler reaction promoted by trimethylchlorosilane / R. Tsuji, M. Nakagawa, A. Nishida // Tetrahedron Asymmetry. – 2003. – Vol. 14, № 2. – P. 177-180.
11. Jiang W. Synthesis of optically pure pyrroloquinolones via Pictet–Spengler and Winterfeldt reactions / W. Jiang, Z. Sui, X. Chen // Tetrahedron Lett. – 2002. – Vol. 43, № 16. – P. 8941-8945.
12. Waldmann H. Asymmetric steering of the Pictet–Spengler reaction by means of amino acid esters as chiral auxiliary groups / H. Waldmann, G. Schmidt, M. Jansen [et al.] // Tetrahedron. – 1994. – Vol. 50, №47. – P. 11865-11884.
13. Kaljuste K. Solid phase synthesis of 1,2,3,4-tetrahydro--carbolines; implications for combinatorial chemistry / K. Kaljuste, A. Unden // Tetrahedron Lett. – 1995. – Vol. 36, № 50. – P. 9211-9214.
14. Connors R. V. The regio- and stereoselective addition of carbon nucleophiles to trifluoromethylphenylsulfanyl acetylene: a novel and expeditious approach to 3-trifluoromethylfurans / R. V. Connors, A. J. Zhang, S. J. Shuttleworth // Tetrahedron Lett. – 2002. – Vol. 43, № 4. – P. 665-667.
15. Solid phase sequential 1,3-dipolar cycloaddition Pictet–Spengler reactions / H. A. Dondas, R. Grigg, W. S. MacLachlan [et al.] // Tetrahedron Lett. – 2000. – Vol. 41, № 6. – P. 967-970.
16. Klein C. Solid-phase synthesis of new fused tetra, penta and hexa-cyclic:-carboline derivatives / C. Klein, J.M. Ostrech, A. Nefzi // Tetrahedron Lett. – 2003. – Vol. 44, № 10. – P. 2211-2215.
17. Hutchins S. M. Solid phase synthesis of tetrahydroisoquinolines&tetrahydroimidazopyridines / S. M.Hutchins, K. T.Chapman // Tetrahedron Lett. –1996. – Vol. 37, № 28. – P. 4865-4868.
18. Циклизации N-гетарил-5-аминопиразолов в реакциях азосочетания и пикте-шпенглера / С. Ю. Зинченко, С. В. Гресько, С. Ю. Суйков [и др.] // Науковіпраці ДонНТУ. – 2008. – № 137(11). – С. 82-92.
19. Богза С. Л. Взаимодействие орто-арилзамещенных аминоазолов с бензальдегидами. / С. Л. Богза // Cтруктура органических соединений и механизмы реакций – 1999. – Т. 2. – С. 25-30.
20. A versatile synthesis of pyrazolo[3,4 - c]isoquinoline derivatives by reaction of 4 � aryl 5- amino-pyrazoles with aryl/heteroaryl aldehydes: the effect of heterocycle on the reaction pathways / S. L. Bogza, K. I. Kobrakov, A. A. Malienko [et.al] // J. Org. Biomol. Chem., 2005. – № 3. – P. 932-940.
21. Богза С. Кислотні циклізації аміноазолів. Синтез поліядерних гетероциклів з фрагментами ізохіноліну та цинноліну / С. Богза, С. Зинченко, С. Суйко // Вісник наукового товариства ім. Шевченка. Донецьке відділення. Хімія, – 2006. – Т. 10. – С. 94-100.
22. Синтез 1,2-транс-арилгликозидов по Гельфериху при катализе ортофосфорной кислотой / Е. Р Новик., В. М. Соколов, Е. П. Студенцов [ и др.] // Журн. Общей химии. – 1986. – Т. 56, вып 1. – С. 181-187.
23. Бочков А. Ф. Образование и расщепление гликозидных связей / А. Ф. Бочков, В. А. Афанасьев. – М.: Наука, – C. 1978. – 180.
24. Konishi F. Synthesis and taste of some flavones and dihydrochalcone glycosides in which carbohydrate moieties are located at differing positions of the aglycones / F. Konishi, S. Esaki, Sh. Kamiya // Agric. Biol. Chem. – 1983. – Vol. 47, № 7. – P. 1419-1429.
25. Tanaka M. The rates of hydrolysis of some substituted phenyl 2-acetamido-2-deoxy-- and –-D-glucopyranozides / M. Tanaka, S. Kyosaka, Y. Ito // Chem. Pharm. Buii. – 1973. – Vol. 21, № 9. – P. 1971-1977
26. Rothermel J. Phase-transfer-catalyzedsynthesisof aryl(-ketosides of N-acetylneuraminicacid / J. Rothermel, H. Faillard //Biochemie. – 1990.– № 190. – P. 29-40.
27. Лазарева Н. В. Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей / Н. В. Лазарева, Э. Н. Левина. – Л.: Химия, 1976. – Т. 1. –. C. 592.
28. Лазарева Н. В. Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей / Н. В. Лазарева, Э. Н. Левина. – Л.: Химия, 1976. – Т. 2. –. C. 624.
29. Лазарева Н. В. Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей / Н. В. Лазарева, Э. Н. Левина. – Л.: Химия, 1976. – Т. 3. – C. 608.
30.Лазарева Н. В. Вредные вещества в промышленности. Справочник для химиков, инженеров и врачей / Н. В. Лазарева, Э. Н. Левина. – Л.: Химия, 1976. – Т. 3. – C. 384.
31. Хираока М. Краун-соединения / М. Хираока. – М.: Мир, –. C. 1986. – 363.
32. Симонович С. В. Компьютер в вашей школе / С. В. Симонович. – М.: Информком-Пресс, –. C. 2001. – 336.
33. Особенности межфазного каталитического гликозилирования салициловой кислоты / Т. А Чупахина., Ю. H. Гончаренко, В. О. Курьянов [и др.] // Ученые записки ТНУ. – 2011. – Т. 24(63), № 2. – С. 396-401.
34. Синтез арил-О--D-глюкозаминидов и оценка их биологической активности в тесте ингибирования биолюминисценции морских светящихся бактерий / В. О. Курьянов, А. М. Кацев, Т. А. Чупахина [и др.] // Журнал орг. та фармхімії. – 2009. – Т. 7, вип. 4(28). – С. 30-40.
35. Чупахина Т. А. Синтез и исследование антимикробной активности глюкозаминидов 8-гидроксихинолинов / Т. А. Чупахина, А. М. Кацев, В. О Курьянов // Биоорган. химия. – 2012. – Т. 38, № 4. – С. 482-488.
36. Синтез гетероароматических S- и N--гликозидов N-ацетилглюкозамина в межфазных условиях / В. О. Курьянов, Т. А. Чупахина, А. Е. Земляков [и др.] // Биоорган. химия. – 2012. – Т. 38, № 4. – С. 482-488.
37. Bruniger H. Benzazolglycoside. IV. Darsterllung von 1--D glucosaminopyranosyl-benzazolen / H. Bruniger, A. Koine // Arch. Pharmaz. und Ber. Dеtsch. pharmaz. Ges. – 1965. – B. 298, № 9. – S. 768-777.
38. Zinner H. Benzazole. XIX. Glycoside des benzthiazolthions / H. Zinner, K. Peseke // Chem. Ber. – 1965. – B. 98, № 11. – S. 3508-3514.
|
ПРИЛОЖЕНИЕ
|
|
|

|
Рисунок 1. 1Н ЯМР спектр соединения 75.
|
|
|
Рисунок 2. 1Н ЯМР спектр соединения 77.
|
|
|
Рисунок 3. 1Н ЯМР спектр соединения 79.
|
|
|
Рисунок 4. 1Н ЯМР спектр соединения 81.
|
|
|
Рисунок 5. 1Н ЯМР спектр соединения 83.
|
Синтез гликозидов N-ацетил-D-глюкозамина с агликонами пиразолоизохинолинов с использованием каталитической межфазной системы «твердое тело – органический растворитель» для изучения их медико-биологические свойства