Рабочие кардиомиоциты млекопитающих
Содержание
Содержание………………………………………………………………………………………2
Введение………………………………………………………………………………………….3
Модели исследования апоптоза кардиомиоцитов……………………………………………..5
у животных………..……………………………………5
у человека..……………………………………………..9
Методы обнаружения апоптоза кардиомиоцитов……………………………………………12
Морфологические признаки апоптоза кардиомиоцитов…………………………………….16
Фармакологическая коррекция апоптоза кардиомиоцитов………………………………….20
Заключение……………………………………………………………………………………...24
Список литературы……………………………………………………………………………..26
Введение
Рабочие кардиомиоциты млекопитающих – это высокодифференцированные, высокоорганизованные клетки, специализированные для выполнения функции сокращения [16]. Кардиомиоциты млекопитающих относят к некамбиальному типу клеточных популяций, что ограничивает регенерационные возможности миокарда. Одной из проблем биологии кардиомиоцитов является их гибель при различных сердечно-сосудистых заболеваниях. В ряде известных к настоящему времени типов гибели клеток [34], безусловно, наиболее изученным остается апоптоз. За последнее время показано, что при многих патологиях сердца, как острых (инфаркт миокарда), так и хронических (сердечная недостаточность) возрастает число кардиомиоцитов с признаками апоптоза [18].
Во многих работах отмечено, что апоптоз – сложный энергозависимый процесс. Это гененетически контролируемый тип клеточной гибели, имеющий отличительные морфологические и биохимические признаки [21]. Апоптоз в норме является одним из важнейших механизмов морфогенеза, антираковой защиты организма, но при этом, в определённых случаях, усиливает тяжёлые поражения организма. В запуске апоптоза участвуют различные органеллы, прежде всего – плазматическая мембрана и митохондрии - органеллы, имеющие особенное значение в функционировании мышечных клеток. Индукция апоптоза и активация проапоптотических белков ведёт к активации каспаз, расщепляющих белки - мишени. В результате происходит разрушение внутриклеточных органелл или их перестройка, фрагментация клетки на апоптотические тельца. Помимо каспазного в последнее время различают некаспазный механизм апоптотической гибели [28], при котором происходит миграция в ядро флавопротеина AIF и эндонуклеазы G, вызывающих распад ядерной ДНК на крупные фрагменты.
Апоптоз кардиомиоцитов довольно редкое событие [17], часто наблюдается вместе с некрозом. При апоптозе, в отличие от некроза, не наблюдается набухания клетки и её органелл, плазматическая мембрана сохраняет свою целостность, не развивается воспалительный процесс. Для идентификации апоптоза кардиомиоцитов применяются различные методы, сравнительный анализ которых является одной из целей данной работы.
Апоптоз может быть индуцирован в кардиомиоцитах в условиях эксперимента (ишемия, стресс, гипоксия и др.) на животных моделях. В клинике при поражениях сердца существует очень мало прямых морфологических доказательств апоптоза кардиомиоцитов, происходящего на всех стадиях инфаркта миокарда, ишемической болезни сердца, несмотря на доступность большинства непрямых доказательств (например, детекция ДНК-фрагментов). Выявление особенностей апоптотической гибели кардиомиоцитов на различных моделях также является целью данной работы.
В настоящее время известно большое число различных фармакологических агентов, способных эффективно ингибировать апоптоз кардиомиоцитов [2,23,27], однако эти вещества пока успешно применяются лишь в экспериментальных условиях. Обсуждение перспектив применения антиапоптотических веществ в клинической практике при лечении сердечно - сосудистых заболеваний занимает важное место в данной работе.
Модели исследования апоптоза кардиомиоцитов
Роль программируемой клеточной гибели в развитии коронарной болезни сердца и ее осложнений, таких как инфаркт миокарда и сердечная недостаточность, привлекает пристальное внимание исследователей [2,7,13]. За последнее время, как на животных, так и у человека получены данные о том, что гибель кардиомиоцитов путём апоптоза играет важную роль при развитии заболеваний сердца.
Модели сердечной недостаточности на животных охватывают большое число видов (крыса, кролик, собака и др.) и условий, в которых проводился эксперимент (ишемия, динамический стресс, аноксия и др.) [1,3,5,6]. Модели можно классифицировать по способам индукции апоптоза, а именно: окклюзия коронарных артерий [2,9,10]; динамический стресс [7]; введение апоптоз-индуцирующих веществ [6]; генетические нарушения [5,8]. Изучение апоптоза, как и других процессов, кардиомиоцитов у человека затруднено, поэтому большинство исследований проводится на животных моделях.
|
Объект
|
Модель
|
Методы детекции
|
|
крыса
|
Аноксия, воздействие изопротеренола, ишемия/реперфузия, стресс – индуцируемая коронарная недостаточность, перерастяжение
|
Электронная микроскопия
TUNEL
Annexin V
Иммунодетекция
(антитела к каспазам)
Электрофорез ДНК
Флуоресцентные красители(Hoechst)
|
|
мышь
|
Динамический стресс, ишемия/реперфузия
|
|
|
кролик
|
Аутоиммунная кардиомиопатия, ишемия/реперфузия
|
|
|
свинья
|
Ишемия/реперфузия
|
|
|
собака
|
Хроническая сердечная недостаточность, микроэмболизация коронарных артерий
|
|
|
человек
|
Ишемическая болезнь, аритмогенная дисплазия правого желудочка, острый инфаркт миокарда
|
|
Изучение апоптоза кардиомиоцитов у животных:
1. Для сердца взрослых крыс характерно развитие массового послеинфарктного апоптоза кардиомиоцитов, приводящее к гибели организма. Исследование повреждающего действия аноксии в основном проводится на клетках из культуры кардиомиоцитов, а не в системе in situ.
В работе В.Б.Сапруновой и соавторов в качестве модели постинфарктного состояния ткани сердца были использованы изолированные кусочки миокарда, которые инкубировались в условиях аноксии (72 часа). После этого было проведено комплексное исследование ряда параметров: дыхание срезов, ультраструктуры митохондрий и межнуклеосомной фрагментации ДНК. Параллельно, в аэробных условиях, на этой модели были исследованы изменения ультраструктуры митохондрий под влиянием TNF- . Проведённые исследования продемонстрировали протекание апоптоза в условиях эксперимента (с помощью методов электронной микроскопии, выделения и электрофореза ДНК) [3].
Одной из причин возрастания числа кардиомиоцитов с признаками апоптоза при различных патологиях сердца может быть повышение активности адренергической системы (действие высоких доз катехоламинов усиливает апоптотическую гибель кардиомиоцитов).
Для индукции апоптоза кардиомиоцитов левого желудочка сердца крыс Л.С.Погодина и соавторы использовали синтетический аналог адреналина – изопротеренол (ИЗП), вызывающего развитие «кальциевого» повреждения митохондрий, впоследствии сочетающегося с ишемией кардиомиоцитов из-за усиленного сокращения сердечной мышцы. Для выявления апоптотических кардиомиоцитов применяли метод TUNEL. Было установлено, что в очагах повреждения присутствуют три вида кардиомиоцитов; показано, что апоптоз преобладает в начале развития очага повреждения (4-18 ч), а на более поздних стадиях (24-48 ч) - скорее всего некроз.[6]
Для изучения роли апоптоза в развитии коронарной болезни сердца Маншарипова А.Т. и соавторы использовали экспериментальную модель стресс-индуцированной коронарной недостаточности. В целях моделирования коронарной недостаточности крысы подвергались стрессовому воздействию в виде длительной иммобилизации животных лёжа на спине в течение 8-ми часов ежедневно на протяжении 11-ти дней. Действие стресса выражается в понижении уровня оксида азота в крови и миокарде. Апоптотические кардиомиоциты определялись методом TUNEL. Было показан массивный характер апоптоза при моделировании коронарной недостаточности (с 3 дня); отмечалась прямая зависимость расширения зоны апоптоза от длительности стрессового воздействия [7].
Также у крыс путём окклюзии левой коронарной артерии моделировали необратимую ишемию, затем исследовали потенциально протекторное действие препарата Семакс. Как и при введении Семакса, так и без него при анализе методом TUNEL не наблюдали апоптотических клеток в миокарде после 2 часов 30 минут, однако без введения данного препарата кардиомиоциты имели значительные нарушения основных органелл. Таким образом, Семакс отсрочил появление патологических изменений в ультраструктуре ишемизированных кардиомиоцитов [9].
H. Fliss и соавторы на моделях ишемии (окклюзия левой коронарной артерии, 2, 25 ч) и ишемии (45 мин) с последующей реперфузией (1-4 ч) показали, что как ишемия, так и реперфузия ведут к апоптотической гибели кардиомиоцитов. В отличие от предыдущей работы Buerke M и соавторов на сердце крыс по изучению действия реперфузии на апоптоз (20 мин. ишемия / 24 ч реперфузия) [15], в данном исследовании было замечено, что развитие апоптоза после реперфузии происходит намного быстрее, чем считалось ранее (достаточно 1 ч реперфузии). Апоптоз кардиомиоцитов оценивали с помощью ISEL метода и «лестницы» ДНК в агарозном геле. По данным исследования реперфузия уменьшает область апоптоза в зоне ишемии миокарда, но, в то же время, она способна усиливать остаточный апоптоз, возможно из-за повреждающего эффекта реперфузии (мощная генерация активных форм кислорода).[10]
Модель механического перерастяжения кардиомиоцитов сосочковых мышц сердца крыс описана Cheng с коллегами, при этом выявлено присутствие апоптотических кардиомиоцитов (21%).
2. Можно выделить ряд работ, в которых проводился анализ гибели кардиомиоцитов у мышей с генетическими аномалиями или у трансгенных животных.
Михайлов В.М. и соавторы использовали в своей работе животных с генетическим дефектом - мышей mdx, у которых нарушение синтеза дистрофина в кардиомиоцитах сопровождалось развитием окислительного стресса, вызывающего в свою очередь гибель клеток. У таких мышей, содержащихся в обычных условиях, имелись признаки развивающегося апоптоза, для включения деструктивной стадии апоптоза, в течение которой развивается низкомолекулярный распад ДНК, необходимо усиление окислительного стресса. Нормальных мышей и мышей mdx подвергали динамическому стрессу – плаванию в холодной воде в течение 5 мин. После динамического стресса были описаны ультраструктурные особенности ядер и митохондрий кардиомиоцитов мышей начальной и деструктивной стадий апоптоза [4], исследована динамика появления и исчезновения двойных разрывов в ядрах кардиомиоцитов (использование антител к фосфорилированной форме гистона -H2AX) [5].
На модели дилатационной кардиомиопатии – у трансгенных мышей с кардиоспецифической активацией каспазы-8 и стерил-20-подобной киназы-1 млекопитающих было показано, что диффузная гибель кардиомиоцитов может обусловливать развитие сердечной недостаточности в отсутствии каких-либо других причин, снижающих сократительную способность кардиомиоцитов [8].
3. На модели ишемии (30 мин) и последующей реперфузии в течение часа сердца у кроликов R.Gottlieb и соавторы показали, что в ответ на реперфузию, но не на ишемию, развивается апоптотическая гибель кардиомиоцитов. Это хорошо согласуется с данными по апоптозу кардиомиоцитов в условиях ишемии на других животных (Fliss). Клиническое значение этих данных состоит в том, что, очевидно, поздняя постинфарктная гибель кардиомиоцитов имеет не некротическую, а апоптотическую природу [2].
4. Апоптоз кардиомиоцитов был индуцирован во время открытой операции на сердце свиней, с помощью аортального зажима создавались условия ишемии. Апоптотические кардиомиоциты определялись методами TUNEL и имунногистохимическим путём связывания с каспазой 3. На данной модели было показано, что с увеличением времени пережима аорты (60 – 90 мин.) апоптотическая гибель кардиомиоцитов усиливается [11].
5. При использовании метода микроэмболизации коронарных артерий у собак с хронической сердечной недостаточностью с помощью электронной микроскопии и иммуногистохимического исследования было обнаружено, что апоптотический тип клеток был зафиксирован не только на границе очагов инфаркта, но и в отдалённых от них участках миокарда. Однако в зоне некроза общая встречаемость апоптотических клеток вне зависимости от гистохимической принадлежности в 3-4 раза превышала фоновый уровень[1].
На той же модели Гусева и соавторы показали, что ранняя длительная терапия с использованием эналаприла снижает апоптоз кардиомиоцитов [12].
Изучение апоптоза кардиомиоцитов у человека:
Для определения роли апоптоза в гибели кардиомиоцитов при ишемической болезни сердца были исследованы сердца умерших больных, страдавших кардиоваскулярной патологией. Для выявления апоптоза использовали метод TUNEL. В результате было показано, что апоптоз является основной формой гибели кардиомиоцитов при ранней стадии ишемии, при крупноочаговом постинфарктном кардиосклерозе и ишемической болезни сердца с острой или прогрессирующей хронической сердечной недостаточностью [13].
В своей работе G.Olivetti и соавторы использовали образцы миокарда 36 пациентов с хронической сердечной недостаточностью (после трансплантации сердца). Апоптоз кардиомиоцитов оценивали биохимически, гистохимически и путём комбинации гистохимического анализа с конфокальной микроскопией, также в данной работе определяли и экспрессию белков BCL2 и BAX. По результатам исследования уровень апоптоза при хронической сердечной недостаточности составил 77%, что отличается от результатов Narula и соавторов (уровень апоптоза составлял от 5% до 35,5%) [18], однако в обеих указанных работах уровень апоптических клеток в миокарде был очень высоким. Также в данном исследовании было показано, что число кардиомиоцитов меченых на белок BCL2 (антиапоптотическая активность) превышало в 1,8 раз контрольный образец, тогда как уровень экспрессии белка BAX оставался постоянным. Таким образом, было показано развитие апоптоза кардиомиоцитов в декомпенсированном сердце человека, несмотря на усилении экспрессии BCL2 [17].
M.Allat и соавторы изучали развитие аритмогенной дисплазии правого желудочка, которая характеризуются интенсивным замещением кардиомиоцитов волокнистой тканью. Было показано, что гибель кардиомиоцитов при этом заболевании происходит путём апоптоза (метод TUNEL) [19].
Хлапов А.П. и соавторы исследовали роль апоптоза кардиомиоцитов в механизмах ишемического ремоделирования миокарда. Материалом исследования являлись биоптаты миокарда левого желудочка, полученные интраоперационно у пациентов с ишемической болезнью сердца и хронической сердечной недостаточностью. Для оценки роли апоптоза кардиомиоцитов в развитии хронической сердечной недостаточности пациенты были разделены на три клинические группы с учетом величин конечного диастолического объема: I группа — менее 120 мл, II — от 120 до 150 мл, III группа — более 150 мл. Показано, что наибольшая встречаемость TUNEL-ядер кардиомиоцитов характерна для I и II клинических групп (0,65 и 0,58 соответственно), то есть была установлена существенная значимость апоптоза кардиомиоцитов на ранних этапах ремоделирования левого желудочка. Таким образом, максимальная эффективность лечебных мероприятий, направленных на терапию ишемического ремоделирования миокарда, может наблюдаться лишь на ранних стадиях заболевания. На более поздних стадиях терапия, направленная на управление апоптозом, может оказаться малоэффективной [14].
В работе A. Abbate и соавторов было показано, что у пациентов умерших (за период 10 дней) после острого инфаркта миокарда постинфарктное ремоделирование миокарда левого желудочка и развитие сердечной недостаточности было связано с апоптозом кардиомиоцитов, причём постинфарктное развитие сердечной недостаточности коррелировало с возросшим приблизительно в 4 раза апоптозом (с 6, 4% до 26, 2%) в области инфаркта. Кроме того апоптоз кардиомиоцитов как в области инфаркта, так и в непоражённых участках усиливал ремоделирование миокарда левого желудочка. Для определения апоптоза кардиомиоцитов использовали метод TUNEL и иммуногистохимический (антитела к каспазе-3) [16].
Методы обнаружения апоптоза кардиомиоцитов
Изначально идентификация апоптических клеток базировалась только на морфологическом критерии [17]. Наличие артефактов и субъективная интерпретация признаков снижало достоверность определения апоптоза в несколько раз, поэтому требовались дополнительные методы.
В настоящее время существует большое количество методов выявления апоптоза кардиомиоцитов: электронная микроскопия, метод TUNEL, окраска Annexin V, иммунодетекция эффекторов апоптоза, электрофорез ДНК, различные флуоресцентные ядерные красители. Использование красителей позволяет зарегистрировать морфологические признаки, такие как конденсация и деградация хроматина. Обнаружение формирующихся разрывов ДНК и ее деградации осуществляется методом электрофореза (выявление “лестницы” ДНК). Наличие олигонуклеотидов, встроенных в ДНК через ее свободные концы, формирующиеся в участках разрывов, определяется с помощью таких морфологических методов как TUNEL (TdT-mediated dUTR-biotin nick end-labeling) или ISEL (in situ end-labeling).
Классификацию методов обнаружения апоптоза кардиомиоцитов можно проводить и по времени появления апоптотических изменений в клетке, так Annexin V метод и иммунодетекцию можно отнести к методам выявления ранних признаков апоптоза (появление фосфатидилсерина на поверхности клетки, активация каспаз), а метод TUNEL, электрофорез ДНК, использование флуоресцентных ядерных красителей, электронная микроскопия – к методам выявления более поздних признаков апоптоза (разрывы ДНК и другие повреждения).
Одним из наиболее распространённых методов детекции апоптоза кардиомиоцитов является TUNEL метод, который позволяет выявлять апоптотические клетки с фрагментацией ДНК, у которых еще отсутствуют или слабо выражены морфологические изменения, характерные для апоптоза. Этот метод основан на применении меченого биотином уридинтрифосфата, который связывается со свободными З.'_ОН концевыми группами фрагментов ДНК. Данный метод обладает высокой надежностью только в случае клеток и тканей с низким пролиферативным индексом, поэтому относительно хорошо подходит для кардиомиоцитов. Необходимо учитывать, что метка будет обнаруживаться также на ДНК в процессе ее репарации, и в некротических тканях [35]. Во время апоптоза под действием эндонуклеаз происходят многочисленные разрывы нитей ДНК, в результате чего образуется множество ее 3'_ОН концов. Их присутствие в клетках можно определить при помощи модифицированных нуклеотидов (например, биотин_dUTP) в реакции мечения однонитевых разрывов, катализируемой ДНК_полимеразой или терминальной дезоксинуклеотидтрансферазой (ТдТ). Меченые 3’_ОН концы молекулы ДНК в индивидуальных клетках затем выявляются с помощью авидина, конъюгированного с флуоресцентным красителем. Этот метод использовали в своей работе Л.С. Погодина и соавторы при индукции апоптоза кардиомиоцитов изопротеренолом [6], А.П. Хлапов и соавторы при исследовании роли апоптоза кардиомиоцитов в механизмах ишемического ремоделирования миокарда [14] и многие другие [7,9,11,13]. Т.Э. Владимирская и соавторы при изучении роли апоптоза кардиомиоцитов в развитии ишемической болезни сердца использовали модифицированный TUNEL метод, в котором визуализация меченых моно - и олигонуклеосомных фрагментов ядерной ДНК осуществлялась путём присоединения вторичных антител, конъюгированных с пероксидазой, и образования иммунокомплексов с субстратом (специальный реагент CytoninTM позволял идентифицировать апоптотические кардиомиоциты) [13].
В настоящее время широко распространён цитофлуориметрический метод выявления апоптотических клеток, однако его применение ограничено при использовании срезов ткани после химической фиксации. ( Проточная цитофлуориметрия – это процесс, в котором измеряются физические и химические характеристики одиночных клеток или других биологических и небиологических частиц приблизительно одинакового размера, проходящие через оптическую систему в потоке жидкости). Одним из основных методов детекции процессов апоптоза с помощью проточной цитофлуориметрии является использование системы ANNEXIN V – FITC , основанной на совместном использовании аннексина V, аффинного к фосфатидилсерину и иодида пропидия, который проникает в клетки, взаимодействуя с ДНК. Фосфатидилсерин – фосфолипид, который в процессе апоптоза локализуется на клеточной поверхности и формирует один из специфических сигналов для распознавания апоптотической клетки. Различное насыщение клетками двумя красителями позволяет определить живые, некротические и апоптотические клетки. Этот метод использовали Dumont E.A и соавторы для идентификации апоптоза кардиомиоцитов, вызванного ишемией / реперфузией [20].
Электрофорез ДНК для выявления апоптоза кардиомиоцитов использовали в своей работе В.Б.Сапрунова и соавторы (индукция апоптоза в условиях аноксии) [3], H. Fliss и соавторы на моделях ишемии и ишемии с последующей реперфузией [10].
Иммуногистохимический метод, а именно антитела к фосфорилированной форме гистона -H2AX, использовали в своей работе Михайлов В.М. и соавторы для регистрации двойных разрывов ДНК (после динамического стресса у нормальных мышей и мышей mdx). Идентификацию апоптоза кардиомиоцитов с помощью антител к каспазе-3 проводили M. Malmberg и соавторы (развитие апоптоза в условиях ишемии с помощью аортального зажима) [11], A. Abbate и соавторы (апоптоз кардиомиоцитов в условиях хронической сердечной недостаточности) [16].
Для определения конденсации и фрагментации хроматина, происходящей при апоптозе, используют флуоресцентные красители: Hoechst, dUTP (deoxyuridine triphosphate), PI (propidium iodide). Например, dUTP и PI применяли G.Olivetti и соавторы при изучении роли апоптоза кардиомиоцитов в развитии ишемической кардиомиопатии у человека.[17]
Для достижения наилучшего результата при идентификации апоптоза кардиомиоцитов используют сразу несколько методов: TUNEL метод и электронная микроскопия [6], электрофорез ДНК и электронная микроскопия [3], TUNEL метод и иммуногистохимия [11].
При интерпретации получаемых результатов следует помнить, что апоптоз является обратимым процессом (до активации каспаз 3, 6, 7), а значит не все клетки, меченные маркерами ранних стадий апоптоза, обязательно погибнут. К примеру, не все кардиомиоциты, аннексин-позитивные и при этом не окрашиваемые пропидий иодидом, а также клетки, окрашиваемые антителами на Fas, обязательно завершат программу апоптоза и погибнут [35].
Морфологические признаки апоптоза кардиомиоцитов
Как неоднократно показано разными группами исследователей, апоптоз кардиомиоцитов (как и других клеток) протекает в две стадии: начальную (медленную) и финальную (быструю). На начальной стадии апоптоза клетка теряет часть цитоплазмы, происходит повреждение ДНК, которое проявляется в конденсации хроматина и появлении инвагинаций ядерной мембраны. Финальная (деструктивная) стадия характеризуется повреждением ядерной оболочки, клеточных мембран, разрушением ядерного содержимого и цитоплазматических органелл [4].
Морфологические признаки ранней стадии апоптоза кардиомиоцитов при использовании соответствующих маркеров видны на электронномикроскопическом уровне, тогда как более поздние апоптотические изменения в структуре кардиомиоцитов можно наблюдать с помощью как электронной, так и световой микроскопии (конденсация хроматина, появление апоптических тел) [21].
Ультраструктурные признаки апоптоза кардиомиоцитов включают:
- Уменьшение объёма апоптотической клетки;
- Инвагинации ядерной оболочки;

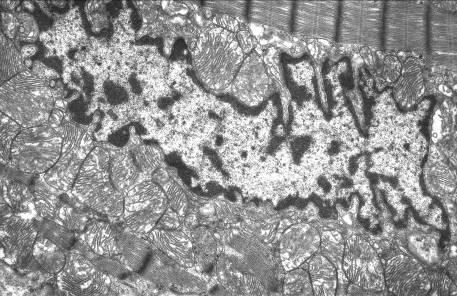
Ультраструктура кардиомиоцита левого желудочка крысы после воздействия ИЗП (Погодина и соавторы, 2006)
- Конденсация хроматина;
- Изменение структуры и положения митохондрий;

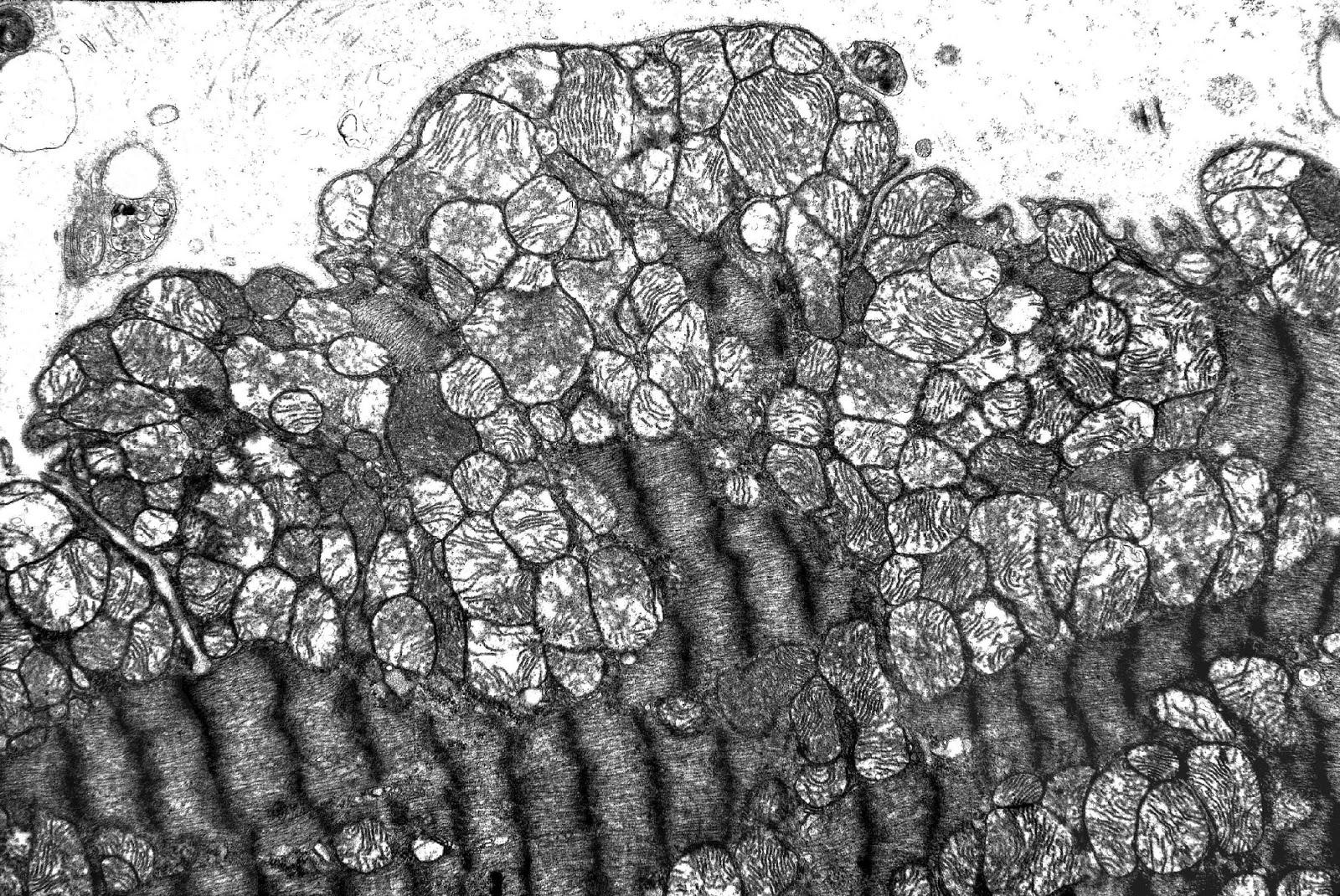
Перемещение митохондрий в кардиомиоците левого желудочка крысы после воздействия ИЗП (Погодина и соавторы, 2006)
- Фрагментация ядра и цитоплазмы с образованием мелких, окружённых мембраной апоптотических тел;
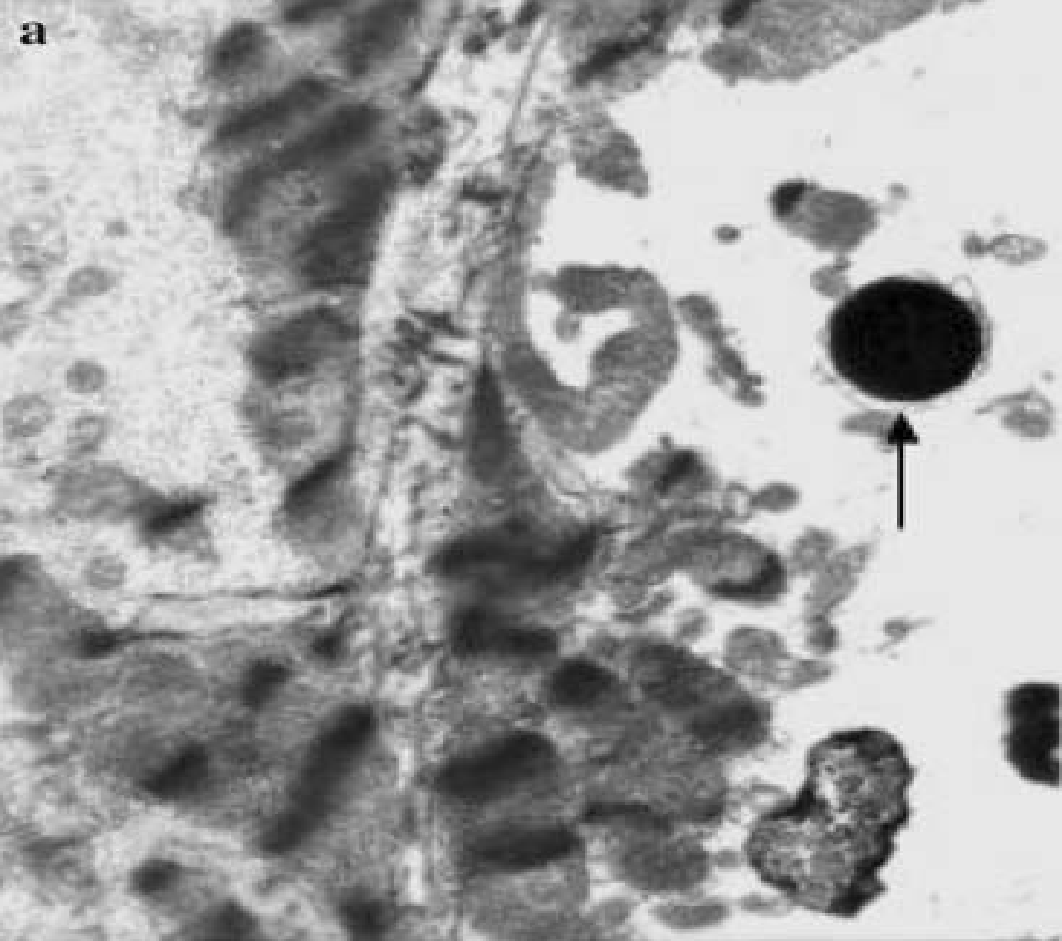
Апоптотическое тело, окружённое фрагментированной мембраной (E.Arbustini и соавторы, 2008)
- Фагоцитоз макрофагами апоптотических тел
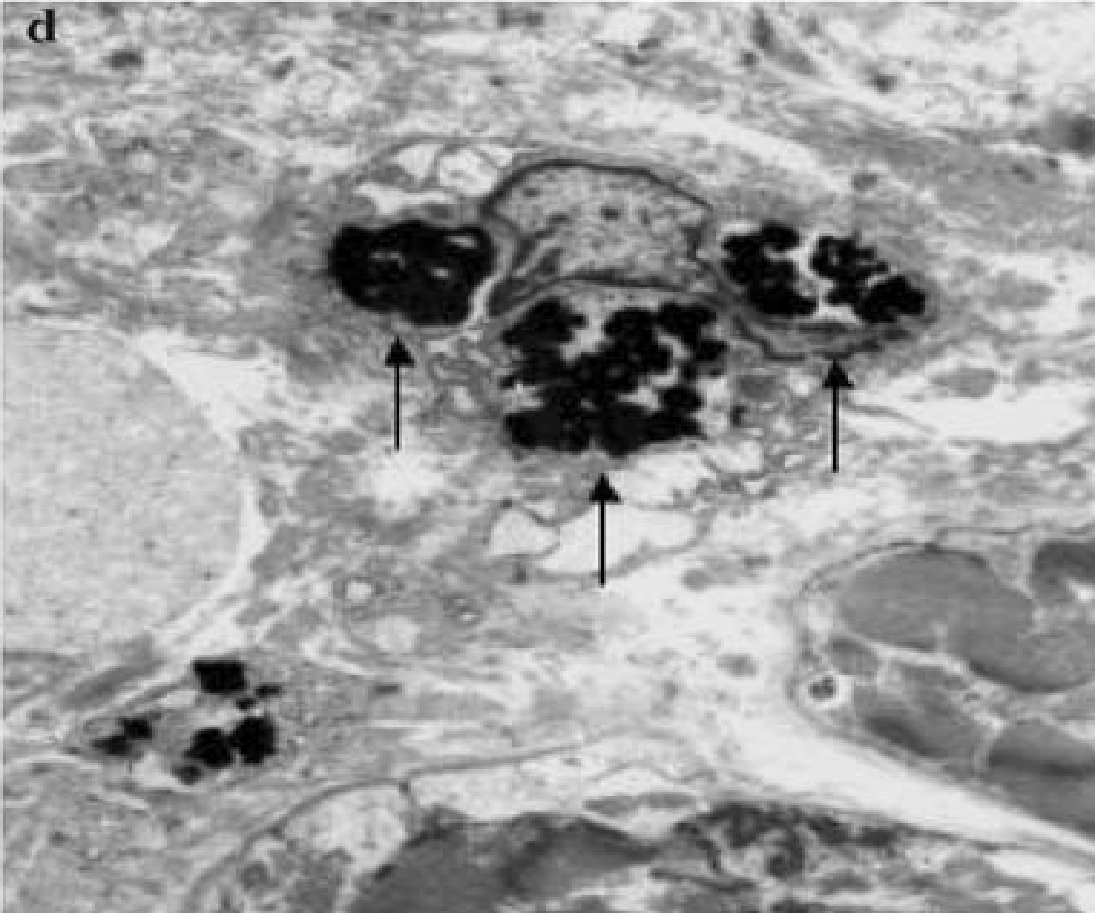
Поглощение макрофагом апоптотических тел (E.Arbustini и соавторы, 2008)
В работе Михайлова В.М. и соавторов были описаны ультраструктурные особенности ядер начальной и деструктивной стадий апоптоза кардиомиоцитов мышей mdx. По степени выраженности инвагинаций ядерной мембраны и распределения конденсированного хроматина ядра кардиомиоцитов мышей mdx были разделены на три типа: нормальные(13%), полупатологические(55%) и патологические(32%). После стресса доля патологических ядер уменьшилась в 2 раза, что объяснялось исчезновением кардиомиоцитов из-за деструктивных стадий апоптоза [4].
Электронномикроскопическое исследование Погодиной Л.С. и соавторов кардиомиоцитов левого желудочка крыс в периферических зонах очагов некроза показало значительную гетерогенность ультраструктуры кардиомиоцитов (различия в форме и структуре ядер, митохондрий, сократительного аппарата). Все кардиомиоциты были разделены на три типа (прежде всего основываясь на различиях в строении ядер): A (овальные ядра с довольно ровной поверхностью и небольшими ядрышками), B (ядра с многочисленными инвагинациями ядерной оболочки, увеличение конденсации хроматина по всему объёму ядра, крупные ядрышки), С (инвагинация ядерной оболочки и конденсация хроматина сильно выражены, ядрышки меньше, чем в контроле) [6].
Митохондрии играют ключевую роль на начальных стадиях развития апоптоза, поэтому изменения в их структуре является важным морфологическим критерием апоптоза кардиомиоцитов. В работе Погодиной Л.С. и соавторов отмечалось просветление матрикса митохондрий с редукцией крист (наиболее выраженное у кардиомиоцитов С типа), также вытеснение митохондрий из центральных участков клетки на периферию, где они скапливались в многочисленных выступах саркоплазмы («почках») [6].
Михайлов В.М. и соавторы также наблюдали уменьшение электронной плотности митохондрий, к тому же были выявлены митохондрии, содержащие овальные включения (кальций-фосфатной природы), что свидетельствовало о том, что гибель кардиомиоцитов происходила не по типу острого ишемического некроза (для начальной, обратимой стадии которого характерно появление игольчатых включений) [4].
При индукции апоптоза кардиомиоцитов в условиях аноксии Сапрунова В.Б. и соавторы наблюдали гетерогенность популяции митохондрий: основную массу составляли митохондрии с обводнённым, просветлённым матриксом, контрастными мембранами, меньшее количество митохондрий характеризовалось сжатым, уплотнённым матриксом. При продолжительном действии аноксии впервые были выявлены локальные перестройки внутренней мембраны митохондрий с образованием ячеистых упорядоченных структур и необычное расположение мелких электронно-плотных митохондрий внутри более крупных с просветлённым матриксом [3].
Фармакологическая коррекция апоптоза кардиомиоцитов.
В настоящее время имеются фармакологические агенты, способные эффективно ингибировать апоптоз кардиомиоцитов, индуцированный различными стимулами: ишемией/реперфузией, H2O2, TNFa и др. Однако эти вещества (ZVAD-fmk, SB 203580, PD 98059, инсулиноподобный ростовой фактор, N-ацетил-цистеин) применяются в основном в экспериментальных условиях [2]. Определённые перспективы связаны с дальнейшим клиническим исследованием карведилола (зарегистрированным фармацевтической фирмой "SmithKleine Beecham Pharmaceuticals" под торговым названием "Coreg(r)"). Препарат представляет собой блокатор - адренорецепторов с выраженной антиоксидантной и умеренной сосудорасширяющей активностью. В проведенных клинических исследованиях карведилол продемонстрировал значительное снижение уровня смертности у больных с сердечной недостаточностью. Механизмом антиапоптотического действия препарата является подавление экспрессии Fas-рецептора на кардиомиоцитах, ингибирование активации SAPK (stress-activated protein kinase – активируемая при стрессе протеин-киназа) [29]. (Fas-опосредованный путь апоптоза также может быть подавлен введением Hsp70 [31]).
Благоприятный эффект даёт применение ингибиторов каспаз. Так, Thomas A. Holly и соавторы использовали в своём исследовании (кролики, 30 минут ишемия/ 4 часа реперфузия) YVAD-cmk (ацетил-Tyr-Val-Ala-Asp - хлорометилкетон), который подавлял активацию каскада каспаз. В результате зона инфаркта сокращалась приблизительно на 31% [30].
В работе Маншарипова А.Т. и соавторов изучалось влияние мицеллярной формы изосорбида динитрата (как экзогенного донора оксида азота) и прогестерона на развитие апоптоза кардиомиоцитов при модели коронарной недостаточности. Проводилось исследование опытной группы животных (крыс), которые подвергались стрессовому воздействию с одновременным введением мицеллярной формы изосорбида динитрата. Было обнаружено снижение количества апоптотических клеток, уменьшение зоны ишемии по сравнению со стрессовой группой животных, также отмечалось улучшение структуры ткани миокарда. При этом ингибирование апоптотического процесса наблюдалось с седьмого дня применения препарата. Исследование срезов опытной группы животных, которым вводили раствор прогестерона одновременно со стрессовым воздействием, показало, что прогестерон также подавляет апоптотическую гибель кардиомиоцитов (вместе с тем при гистологическом исследовании миокарда отмечалась волнообразная деформация кардиомиоцитов и нарушение структуры миокарда).
Таким образом, в данной работе было показано на фоне иммобилизационного стресса ингибирующее действие мицеллярной формы изосорбида динитрата и прогестерона на апоптотические изменения кардиомиоцитов. Авторы данного исследования предполагают, что механизмы подавления апоптоза связаны со сложным взаимодействием между белками теплового шока, антиапоптотическими белками Bcl_2, различными каспазами и оксидом азота. На основании проведённых экспериментов был сделан вывод, что экзогенный донор оксида азота - мицеллярная форма изосорбида динитрата помимо сосудорасширяющего действия может оказывать ингибирующее действие на апоптоз и уменьшать зону ишемии. Иными словами, данный препарат может быть рекомендован для применения в клинической практике для проведения направленного фармакологического воздействия на развитие апоптотических изменений в миокарде при коронарной болезни сердца и сердечной недостаточности [7].
В последнее время интенсивно изучается возможность применения эритропоэтина (гликопротеинового гормона, регулирующего эритропоэз) в клинической практике [23] . Одним из плейотропных эффектов (лечение анемии, улучшение сердечной функции, нейропротекция) эритропоэтина (ЭПО) и его аналогов (например, РЭПО – рекомбинантный эритропоэтин) является снижение апоптоза кардиомиоцитов при различных заболеваниях сердца (возможно из-за увеличения экспрессии Hsp70 и минимизации NF-kB в миокарде после введения ЭПО).
Parsa C.J. и соавторы показали, что предварительная обработка ЭПО миобластов, выделенных из сердец крыс, уменьшает перекисный апоптоз приблизительно на 50%, защищает миобласты от повреждения в течение 12 ч аноксии . Также авторы изучали эффект ЭПО in vivo, вводя его кроликам до индуцирования инфаркта миокарда. Через 6 ч после инфаркта в группе ЭПО выявлялось гораздо меньше апоптотических клеток (18.5%), чем в группе контроля, получавшей физиологический раствор (24%). Через 3 дня наблюдалось сокращение зоны инфаркта в группе ЭПО с 35.1% до 13.8% [22].
L. Calvillo и соавторы исследовали культуру кардиомиоцитов крыс и показали, что ЭПО предотвращает апоптоз кардиомиоцитов, подвергнутых 28 часовой гипоксии. При экспериментальном инфаркте миокарда введение РЭПО (5,000 МЕ/кг веса внутрибрюшинно ежедневно в течение 7 дней) уменьшало повреждение кардиомиоцитов на 50% и нормализовало гемодинамику в течение 1 недели после реперфузии [24].
В другом экспериментальном исследовании на крысах (окллюзионная ишемия продолжительностью 45 минут с последующей реперфузией) Lipsic E и соавторы выявили значительное снижение апоптоза кардиомиоцитов на фоне лечения ЭПО (5,000 МЕ/кг) сразу после ишемии (снижение 29%) и после реперфузии (38%) [25].
Кардиопротекторное действие IGF-I (инсулиноподобный фактор роста) изучалось в работе M.Buerke и соавторами на модели ишемии(20 минут)/реперфузии у крыс. Введение этого фактора роста демонстрировало снижение уровня апоптоза кардиомиоцитов с 62% до 28% [26].
В другой работе также на модели ишемии (45 минут) / реперфузии у крыс исследовалось влияние беназеприла (ингибитора ангиотензин–превращающего фермента) на миокард. Было показано, что беназеприл оказывает антиапоптотическое действие, усиливая экспрессию антиапоптотического белка BCL2 и подавляя экспрессию проапоптотического белка BAX [27].
Применение различных гипотензивных средств (иналаприл, лозартан) также эффективно в предотвращении апоптоза кардиомиоцитов [32].
Таким образом, апоптоз кардиомиоцитов может быть ингибирован путём подавления факторов, участвующих в запуске этого процесса (ингибиторы каспаз, Fas-рецепторов), усилением активности антиапоптотических белков (беназеприл). Существует большое разнообразие веществ, обладающих антиапоптотическим действием на кардиомиоциты, что открывает новые возможности в лечении различных заболеваний сердца. Но проблема в том, что до сих пор до конца не ясно, сможет ли ингибирование апоптоза кардиомиоцитов предотвратить или прекратить развитие, например, сердечной недостаточности. К тому же большинство описанных антиапоптотических агентов изучено пока только на экспериментальных животных моделях; не исследовалась безопасность их применения для других систем организма (ведь апоптоз необходим для нормального функционирования, например, иммунной системы). Более того, стало известно [33], что почти все эффекторные молекулы апоптоза выполняют в клетке и другие функции, не связанные с клеточной гибелью (например, адаптивный ответ на стресс), и в случае нерационального подавления таких молекул могут возникнуть нарушения жизненно-важных функций клетки.
Заключение
В последнее время апоптоз кардиомиоцитов является предметом интенсивного изучения как один из важных факторов в развитии целого ряда заболеваний сердца. Изучение апоптоза, как и других процессов, кардиомиоцитов у человека затруднено, поэтому большинство исследований проводится на животных моделях. В этих случаях индукцию апоптоза кардиомиоцитов осуществляют следующими способами: окклюзия коронарных артерий, динамический стресс, введение апоптоз-индуцирующих веществ, перерастяжение клеток, создание генетических моделей.
Существуют данные и по апоптозу кардиомиоцитов у человека. Присутствие кардиомиоцитов с признаками апоптоза обнаружено у пациентов с острым инфарктом миокарда, ишемической болезнью сердца, хронической сердечной недостаточностью, аритмогенной дисплазией правого желудочка. Во всех случаях уровень апоптоза был высоким (5% - 35.5%), что согласуется с результатами, полученными на животных моделях (0.6% - 33%).
Апоптотические кардиомиоциты млекопитающих обладают рядом отличительных морфологических признаков, главными из которых являются: инвагинации ядерной оболочки; конденсация хроматина (отсутствуют характерные для апоптотических клеток других типов полулуния гетерохроматина); изменение структуры и положения митохондрий (смещение на периферию); фрагментация ядра и цитоплазмы с образованием апоптотических тел (редко описываемых на ткани сердца).
Идентификация апоптоза кардиомиоцитов осуществляется разнообразными методами (электронная микроскопия, метод TUNEL, Annexin V, иммунодетекция, электрофорез ДНК, различные флуоресцентные ядерные красители), наиболее часто используемым можно назвать TUNEL. Наилучший результат даёт применение сразу нескольких методов.
Апоптоз кардиомиоцитов может быть эффективно ингибирован веществами с различным механизмом действия (карведилол, эритропоэтин, беназеприл и др.) Но влияние большинства из них изучено ещё недостаточно, что затрудняет их использование в клинической практике.
Список литературы
- Sharov V.G., Sabbah H.N., Shimoiama H. et al. Evidence of Cardiocyte Apoptosis in Myocardium of Dogs with Chronic Heart Failure // Am J Pathol 1996; 148 (1): 41-9.
- Сторожаков Г.И., Утешев Д.Б. Роль апоптоза в развитии атеросклероза, ишемии миокарда и сердечной недостаточности // Сердечная недостаточность. Оригинальные статьи. 2000. Т.1. №4
- Сапрунова В.Б., Казимирчук С.А., Тоньшин А.А., Бакеева Л.Е., Ягужинский Л.С. Индукция апоптоза в миокарде крыс в условиях аноксии // Биохимия. 2002. Т. 67. С. 293–302.
- Михайлов В.М., Комаров С.А., Нилова В.К., Штейн Г.И., Баранов В.С. Ультраструктурный и морфометрический анализ стадий апоптоза кардиомиоцитов мышей mdx // Цитология. 2001. 43(8):729-737.
- Михайлов В.М., Веженкова И.В. Двунитевые разрывы ДНК кардиомиоцитов мышей C57BL и mdx после динамического стресса // Цитология. 2007. 49(6): 491-496.
- Погодина Л.С., Шорникова М.В., Ченцов Ю.С. Электронномикроскопическая характеристика кардиомиоцитов левого желудочка сердца крыс после индукции апоптоза изопротеренолом // Известия РАН. Серия биологическая. 2006, № 1, с.26-37.
- Маншарипова А.Т., Джусипов А.К., Берсимбаев Р.И., Абылайулы Ж.А., Югай Е.Э., Булентаева З.А. Денисов Ю.Д. Феномен апоптоза и его регуляция при модели коронарной недостаточности // Кардиология СНГ. 2005. Т.3. С.29-32
- Лушникова Е.Л., Непомнящих Л.М., Розенберг В.Д. Морфологические и молекулярно-генетические основы дилатационной кардиомиопатии // Издательство РАМН. 2004. С.192.
- А.В.Голубева, С.А. Гаврилова, Т.В. Липина, М.В. Шорникова, А.Б. Постников, Л.А. Андреева, Ю.С. Ченцов, В.Б. Кошелев. Защитное действие пептида семакс в острую стадию ишемии миокарда крыс // Российский физиологический журнал (РФЖ им. И.М. Сеченова). 2006. Т. 92, № 6, C. 732-745.
- Fliss H., Gattinger D. Apoptosis in Ischemic and Reperfused Rat Myocardium // Circulation Research. 1996; 79(5):949 -956
- Markus Malmberg ,Tommi Va.ha.silta , Antti Saraste , Ville Kyto, Jan Kiss , Erkki Kentala , Markku Kallajoki , Timo Savunen. Cardiomyocyte apoptosis and duration of aortic clamping in pig model of open heart surgery // European Journal of Cardio-thoracic Surgery. 2006. 30. 480—484.
- Anastassia Goussev, Victor G. Sharov, Hisashi Shimoyama, Mitsuhiro Tanimura, Michael Lesch, Sidney Goldstein and Hani N. Sabbah. Effects of ACE inhibition on cardiomyocyte apoptosis in dogs with heart failure // Am J Physiol Heart Circ Physiol.1998. 275:626-631.
- Владимирская Т.И., Швед И.А., Хулуп Г.Я. Верификация роли апоптоза в гибели кардиомиоцитов при ишемической болезни сердца // Здравоохранение; научно - практический ежемесячный журнал / Министерство здравоохранения Республики Беларусь. 2008, №8. С.14-16.
- Хлапов А.П., Вечерский Ю.Ю., Рязанцева Н.В., Калюжин В.В., Мустафина Л.Р., Шипулин В.М., Новицкий В.В. Роль апоптоза кардиомиоцитов в механизмах ишемического ремоделирования миокарда // Бюллетень сибирской медицины.2008, № 3. С.33-38.
- Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion // Proc Natl Acad Sci U S A. 1995; 92:8031-8035.
- Antonio Abbate, Giuseppe G. L. Biondi-Zoccai, Rossana Bussani, Aldo Dobrina, Debora Camilot, Florinda Feroce, Raffaele Rossiello, Feliciano Baldi, Furio Silvestri, Luigi M. Biasucci, Alfonso Baldi. Increased Myocardial Apoptosis in Patients With Unfavorable Left Ventricular Remodeling and Early Symptomatic Post-Infarction Heart Failure // J Am Coll Cardiol 2003; 41:753–760
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, et al. Apoptosis in the failing human heart // N Engl J Med. 1997; 336:1131–1141.
- Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in endstage heart failure // N Engl J Med 1996; 335:1182-9.
- Mallat Z, Tedgui A, Fontaliran F, Frank K, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia // N Engl J Med 1996; 335 : 1190-6.
- Dumont EA, Hofstra L, van Heerde WL, van den Eijnde S,Doevendans PA, DeMuinck E, Daemen MA, Smits JF,Frederik P, Wellens HJ, Daemen MJ, and Reutelingsperger C.P. Cardiomyocyte death induced by myocardial ischemia and reperfusion: measurement with recombinant human annexin-V in a mouse model // Circulation.2000. 102:1564–1568
- Eloisa Arbustini, Agnese Brega, Jagat Narula. Ultrastructural definition of apoptosis in heart failure // Heart Fail Rev. 2008. 13:121–135
- Parsa C.J., Matsumoto A., Kim J. et al. A novel protective effect of erythropoietin in the infarcted heart // J Clin Invest. 2003; 112: 999-1007.
- Бакшеев В.И., Коломоец Н.М. Эритропоэтин в клинической практике: прошлое, настоящее и будущее // Клиническая медицина. 2007.
- Calvillo L., Latini R., Kajstura J. et al. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling // Proc Natl Acad Sci U S A. 2003; 100(8):4802-4806.
- Lipsic E., van der Meer P., Henning R.H. et al. Timing of erythropoietin treatment for cardioprotection in ischemia/reperfusion // J Cardiovasc Pharmacol. 2004; 44(4):473-479.
- Buerke, M, et al. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion // Proc Natl Acad Sci USA 1995. 92:8031-8035.
- Kanhei Charan Sahoo et al. Cardioprotective effects of benazepril, an angiotensin-converting enzyme inhibitor, in an ischaemia-reperfusion model of myocardial infarction in rats // J Renin Angiotensin Aldosterone Syst 2009; 10; 201-209.
- Chen M. and Wang J // Apoptosis.2002. 7:313-319
- Yue TL, Ma XL, Wang X, Romanic AM, Liu GL, Louden C, Gu JL, Kumar S, Poste G, Ruffolo RR Jr, et al. Possible involvement of stressactivated protein kinase signaling pathway and Fas receptor expression in prevention of ischemia/reperfusion-induced cardiomyocyte apoptosis by carvedilol // Circ Res. 1998; 82:166 –174.
- Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ, Cryns VL. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo // J Mol Cell Cardiol. 1999; 31:1709–1715.
- Yun Zhao, Wanyin Wang, and Lingjia Qian. Hsp70 may protect cardiomyocytes from stress-induced injury by inhibiting Fas-mediated apoptosis // Cell Stress & Chaperones. 2007. 12 (1):83–95.
- Fortuno M.A., Gonzalez A., Ravassa S. et al. Clinical implications of apoptosis in hypertensive heart disease // Am. J. Physiol. Heart Circ. Physiol. 2003. V. 284. P. H1495—H1506.
- Galluzzi L., Joza N., Tasdemir E., Maiuri M.C., Hengartner M., Abrams J.M., Tavernarakis N., Penninger J., Madeo F. & Kroemer G. No death without life: vital functions of apoptotic effectors // Cell Death and Differentiation. 2008. 15. 1113-1123.
- G Kroemer, L Galluzzi, P Vandenabeele, J Abrams, E S Alnemri, E H Baehrecke, M V Blagosklonny, W S El-deiry, P Golstein, D R Green. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009 // Cell Death and Differentiation. 2009. 16. 3-11
- Evaristo Castedo et al. Influence of hypothermia on right atrial cardiomyocyte apoptosis in patients undergoing aortic valve replacement // Journal of Cardiothoracic Surgery 2007, 2-7.
- Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonneblick EH, Hintze TH, Olivetti G, Anversa P. Stretch-induced programmed myocyte cell death // Clin Invest.1995. 96(5):2247-59.
PAGE1
Рабочие кардиомиоциты млекопитающих