Влияние физических и химических факторов на основность алкиламинов
Страница 3
Существенные различия между свойствами в газовой и конденсированной фазах наблюдается и при сравнении оснований одного и того же класса. Например, все первичные алкиламины в газовой фазе (№2—29), за исключением b,b,b-трифторэтиламина (№30), оказались более основными, в то время как в воде (см. например, табл. 1) амины с электроотрицательными заместителями зачастую менее основны, чем аммиак. То самое относится и ко вторичным и третичным алкиламинам.
Данные по изменению свободной энергии и энтальпии реакций, описываемых уравнениями (17) — (2), совместно с некоторыми другими результатами позволили определить термодинамические характеристики процессов переноса свободных и протонированпых оснований из газовой фазы в водные растворы и на этой основе про вести термодинамический анализ влияния сольватации на основность аминов в воде [3, 6, 47, 140, 151, 153]. При этом преимущественное внимание было уделено причинам, обусловливающим наблюдаемый порядок изменения основностии в воде при переходе от аммиака к первичным, вторичным и третичным алкиламинам с насыщенными углеводородными заместителями. На основе этих данных Ариетт с сотрудниками [3, 6, 47] сделал вывод, что главным фактором, определяющим наблюдаемый порядок основности аминов различных классов в воде, является специфическая сольватация соответствующих катионов, зависящая от числа атомов водорода у протонированного азота. Неспецифическая же сольватация, по их мнению, имеет второстепенное значение, т. е. эти исследователи придерживаются сольватационной (гидратационной) теории Тротмана — Диккенсона (см. выше).
В то же время другая группа исследователей [140, 153] считает, что изменение основности аминов при переходе из газовой фазы в воду обусловлено прежде всего электростатической (неспецифической) сольватацией катионов, а специфическое взаимодействие играет второстепенную роль. При этом в указанных работах принимается, что кислотно-основные свойства соединений в газовой фазе являются истинными (собственными) свойствами, и в противоположность случаю в конденсированной фазе практически не обсуждается зависимость этих свойств от строения аминов.
Следует отметить, что, несмотря на большой интерес, проявляемый к результатам по основности аминосоединеиий в газовой фазе, пока еще нет общего подхода к объяснению эффектов их структуры на данное свойство. Выявлены только некоторые закономерности, характеризующие поведение отдельных групп родственных аминов. Например, Тафт рассмотрел изменение основности при переходе от аммиака к первичным, вторичным и третичным аминам и нашел, что введение одной, двух или трех алкильных групп (СН3; С2Н5; n-С3Н7; Н2С=СН—СН2; НСЕºС—СН2) сопровождается ростом величин DGB в соотношении 1,00 : 1,72 : 2,22. Повышающее основность действие метильных групп при последовательном накоплении их в a-положении может быть представлено пропорциональностью 1,00: :1,85 : 2,60 [7]. Введение метильной группы в a-положение увеличивает основность амина примерно на 2,1 ккал/моль, в b-и g-положения — на 0,9 и 0,5 ккал/моль соответственно [155].
При сопоставлении ароматических и алифатических аминов с одинаковым числом углеродных атомов у атома азота было найдено, что изменение гибридизации a-атомов углерода (например, переход от пиридина к N-метилпирролидину, от анилина к циклогексиламину) практически одинаково влияет на изменение основности в воде и газовой фазе [7]. Была также обнаружена приблизительно прямолинейная зависимость между изменениями основности аминосоединений, имеющих одинаковое число углеродных атомов у азота, но разный характер гетероатома, и степенью этой гибридизации [159], а также между основностью алкиламипов и степенью гибридизации b-углеродного атома в алкильном радикале [7]. В тех случаях, когда варьирование заместителя происходит не у реакционного центра, были выявлены более строгие закономерности влияния структуры на основность аминов. Так, найдена корреляция между DGB для a-замещенных триметиламинов и sI этих заместителей [7]. Величины DGB для 3-й 4-замещенных пиридинов хорошо коррелируют [7, 162, 163] с их основностью в воде и с постоянными sI (s°) и sR+ (sG+) характеризующими электронные эффекты заместителей [7, 158, 163]. Аналогичные зависимости (но менее строгие) можно получить и при подобных сопоставлениях основности замещенных анилинов в газовой фазе [3, 7].
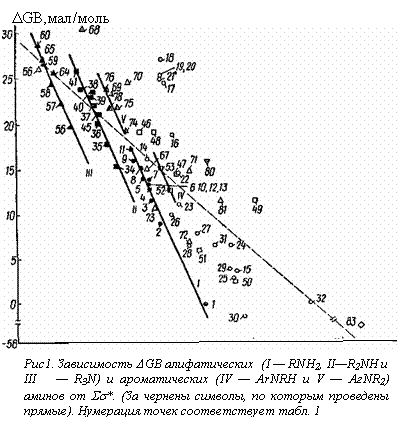 Непосредственное сравнение величин DGB (см. табл. 1) со значениями Ss* заместителей*, присоединенных к атому азота, показывает что, на первый взгляд, здесь отсутствует какая-либо зависимость. Тем не менее имеется некоторая тенденция к уменьшению основности рассматриваемых соединений с ростом электро-отрицателыюсти заместителей в них. Это позволило через 40 (из 47) точек для различных аминосоединений (алкиламины, ариламины, производные гидразина и амиды), основности которых в газовой фазе были известны к концу 1974 г., провести прямую, описываемую [158] уравнением**
Непосредственное сравнение величин DGB (см. табл. 1) со значениями Ss* заместителей*, присоединенных к атому азота, показывает что, на первый взгляд, здесь отсутствует какая-либо зависимость. Тем не менее имеется некоторая тенденция к уменьшению основности рассматриваемых соединений с ростом электро-отрицателыюсти заместителей в них. Это позволило через 40 (из 47) точек для различных аминосоединений (алкиламины, ариламины, производные гидразина и амиды), основности которых в газовой фазе были известны к концу 1974 г., провести прямую, описываемую [158] уравнением**
DGB = (2,1±0,1) — (6,46±0,16)Ss*, (s = 2,1; r = 0,988). (3)
Если аналогичную прямую (пунктирная линия на рис. 4) провести через каждую 71 точку, представленную на указанном рисунке, то ее уравнение имеет вид
DGB = (23,9 ± 0,7) — (8,94 ± 0,48) Ss*,
(s = 4,73; r =0,914). (4)
Следует отметить, что в этом случае при сравнительно узких доверительных интервалах в параметрах уравнения (20) на рис. 4 наблюдаются довольно значительные отклонения от указанной прямой/
Например, точка для аммиака (№ 1) отклоняется вниз, а для тетра-метилендиамина (№ 18) — вверх почти на 11 ккал/моль. Более того, и так невысокий (0,914) коэффициент корреляции значительно уменьшается (до 0,798) при исключении из рассмотрения далеко отстоящей точки для №3 (№ 83). Поэтому найденную зависимость (уравнение (4)), вероятно, можно рассматривать как качественное соотношение, отражающее указанную выше тенденцию к уменьшению DGB с ростом электроноакцепторности заместителей в аминосоединеииях.
Интересные результаты получаются при рассмотрении величин DGB для алкиламинов с насыщенными углеводородными радикалами. Как видно из рис. 4, соответствующие точки (полностью зачерненные символы) группируются таким образом, что для первичных, вторичных и третичных аминов можно провести отдельные прямые [164] с наклонами, соответственно равными: —22,8 ± 2,2; —23,9 ± 2,7 и —23,5 ± 2,2.
Наличие отдельных прямых для алкиламинов различных классов не является неожиданностью. Так, при подобной обработке (сопоставление с Ss*) потенциалов ионизации — одной из важнейших составляющих сродства к протону в газовой фазе [47, 151, 153]) — было найдено [165], что по аналогии с корреляцией потенциалов ионизации различных органических соединений RxМНy (где М =С, О и S) эти данные лучше всего представлять в виде отдельных зависимостей для первичных, вторичных и третичных аминов, хотя имеется и другой подход, в соответствии с которым зависимость потенциалов ионизации аминов от их структуры описывается единым уравнением [166]. Однако первый подход более предпочтителен, поскольку он охватывает больший набор аминов, а также рассматривает с единых позиций потенциалы ионизации самых различных соединений*. Кроме того, при сопоставлении величин РА с потенциалами ионизации [153, 155] и энергиями связывания остовных (1s) электронов [167] наблюдается также отдельные прямые для разных классов аминов. Следует отметить, что при сравнении термодинамических характеристик процессов протонирования аминов в газовой и конденсированной фазах, общей и электростатической теплот гидратации алкиламмоний-ионов как с величинами РА, так и с радиусами этих ионов были получены отдельные прямые для первичных, вторичных и третичных аминов [3, 140, 153]. При этом амины с электроотрицательными заместителями в тех случаях, когда соответствующие данные рассматривались, заметно отклонялись от найденных зависимостей [140]. Из рис. 1 отчетливо видно, что точки (незачерненные символы) для аминов, содержащих электроотрицательные заместители, отклоняются (иногда существенно) от полученных прямых, т.е. здесь наблюдается то же явление, что и при сопоставлении величин DН протонирования аминов в воде и газовой фазе.