Влияние физических и химических факторов на основность алкиламинов
Страница 6
Понятие “индуктивный эффект” достаточно хорошо обосновано экспериментально и является очень полезным для химиков-органиков, однако стало очевидным, что алкильные группы способны стабилизовать не только положительные, но и отрицательные ионы. Это следует из характера кислотности спиртов в газовой фазе (ВuОН > EtOН > МеОН > Н2О) и влияния алкильных заместителей на увеличение силы кислоты в растворе. Этот эффект можно представить как результат делокализации .положительного или отрицательного заряда в молекуле вследствие поляризации различных связей. Как и следует ожидать, .электроотрицательные атомы понижают основность, например, в ряду:
СН3СН2NН2 > FСН2СН2NH2 > F2СНСН2NН2 > F3ССН2NH2
Оценка основности в газовой фазе относительно аммиака
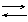 NH4 + B NH3 + BH
NH4 + B NH3 + BH
DG0 = GB(NH3) - GB(В)
Таблица 2. Основность некоторых аминов в газовой фазе
|
соединение |
DG0, кДж/моль |
соединение |
DG0, кДж/моль |
|

|
-101,7 |

|
-75,0 |
|

|
-103,0 |

|
-46,9 |
|

|
-84,2 |

|
-64,9 |
|

|
-84,6 |
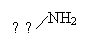
|
-28,1 |
|

|
-75,4 |
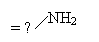
|
-47,3 |
|
|
|
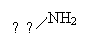
|
-54,4 |
дала возможность сравнить циклические и ациклические амины (табл. 1); важно отметить, что для аминов, имеющих аналогичные заместители, значения DG0 близки (отрицательные значения указывают на большую силу основания). Отклонение, наблюдаемое в случае азиридина, находит объяснение в рамках теории гибридизации: орбиталь свободной электронной пары в азиридине носит более выраженный s-характер, чем в диметиламине, и поэтому менее вероятно ее участие в образовании связи с протоном. Для бензиламинов, по-видимому, экспериментальных данных недостаточно; для трех первичных аминов, представленных в табл. 1, наблюдается удовлетворительная корреляция между характером гибридизации b-углеродного атома и значением DG0 .
Закономерности, выявленные для основности различных аминов в газовой фазе, привлекают своей простотой и четкостью. Большинство экспериментов в органической химии осуществляется в растворах, и в этих случаях изменение основности может иногда описываться приблизительно такими же закономерностями, как и в случае газовой фазы (например, Bu3 > Вu2NН > ВuNН2, полученный для растворов в хлорбензоле относительно 2,4-динитрофенола). Однако часто эти выводы не носят общего характера; так, в бензоле наблюдается следующий порядок основности: Вu2NН > Вu3Н > ВuNH2. В течение многих лет химики проявляли особый интерес к закономерностям, существующим в водных растворах. В этом случае основным параметром является свободная энергия протонирования основания в воде DG0 (Н2О), выражаемая обычно как рKа сопряженной кислоты +ВН [DG0 (Н2О) = —RТlnКа] - Значения рKa для простейших аминов приведены в табл. 2.
Таблица 2. Значение рКа кислот, сопряжённых с алкиаминами (Н2О, 25 0С)
|
Соединение |
рКа |
|
R=Et |
R=Me |
|
R3N |
10,85 |
9,80 |
|
R2NH |
11,09 |
10,73 |
|
RNH2 |
10,80 |
10,66 |
|
NH3 |
9,25 |
Отсутствие четкой закономерности в поведении алкиламинов, объясняли по-разному. Влияние пространственных факторов на стадии протонирования можно не учитывать, и долгое время признавалась важность эффектов сольватации, протекающей в различной степени. Недавно Ауэ применил эти данные в сочетании с известными термодинамическими параметрами в водных растворах для всестороннего анализа дифференциальной сольватации [142]. В ряду алкиламинов теплоты гидратации обычно закономерно понижаются с увеличением размеров молекул. Это влияние алкильных заместителей, называемое гидрофобными эффектами, изучено недостаточно, однако предполагают, что подобные эффекты почти полностью отсутствуют в нейтральных и протонированных аминах, находящихся в водной системе. Считают, что в растворе важным фактором является влияние на сопряженную кислоту ослабления взаимодействия между растворителем и протонированным амином при делокализации заряда в ионе. К тем же выводам приходят при интерпретации этого явления с точки зрения электростатической сольватации (считают, что энергия сольватации и ионный объем связаны обратной зависимостью) и сольватации с участием специфических водородных связей (при этом каждая специфическая водородная связь ослабляется вследствие делокализации положительного заряда в ионе). Таким образом, в тех случаях, когда усиление поляризуемости вследствие увеличения числа алкильных заместителей приводит к стабилизации иона аммония за счет делокализации заряда, сольватация иона должна происходить менее экзотермично, способствуя ослаблению стабилизующего влияния заместителей по сравнению с тем, что имеет место в газовой фазе. Поэтому в ряду алифатических аминов суммарное влияние увеличения степени алкилирования постепенно ослабевает и может фактически приводить к обращению ряда в тех случаях (например, Ме3N в табл. 2), когда эффект уменьшения стабилизации при сольватации сильнее, чем внутримолекулярное стабилизующее влияние алкильных заместителей. И, наоборот, в тех случаях, когда индуктивные эффекты могут вызывать дестабилизацию иона аммония, ион будет обладать повышенной плотностью заряда на атоме азота и лучше сольватироваться; здесь вновь наблюдается противодействие электронным эффектам. Важность сольватации можно подчеркнуть тем, что изменение свободной энергии при переходе ионов аммония из газовой фазы в водный раствор может составлять до 25— 110 кДж/моль (примерно аналогично изменению DG0 за счет электронных эффектов алкильных заместителей в газовой фазе). Для более подробного и систематического знакомства с термодинамическим аспектом данной проблемы и уяснения природы эффектов сольватации читателю следует обратиться к работам [140—142].