Секвенирование (Генная инженерия)
Страница 2

Рисунок 5 Использование ферментативного синтеза олиго(поли)дезоксирибонуклеотидов для определения первичной структуры ДНК.
завершающем этапе анализа. Инкубационную смесь 32Р-меченных олигонуклеотидов различной длины в виде комплексов с матричным полинуклеотидом подвергают гель-фильтрации для удаления дезоксирибонуклеозид-5'-трифосфатов и аликвоты реакционной смеси реинкубируют с ДНК-полимеразой в различных условиях. В случае "минус-системы" реинкубацию проводят в присутствии только трех дезоксирибонуклеозид-5'-три-фосфатов. Например, в «- А-системе» отсутствует dATP и каждая копия в смеси достраивается с помощью ДНК-полимеразы до места, в котором следующим мономерным звеном должен быть остаток pdA (т.е. Т в матричном полинуклеотиде). Образующуюся смесь фракционируют с помощью электрофореза и обнаруживают (ауторадиографически) ограниченное количество полос (их количество равно количеству Т в матричном полинуклеотиде). Аналогично проводят копирование в отсутствие других субстратов: - dGTP, - dCTP или - dTTP (- G-,- С- и - Т-системы соответственно). Все четыре ионофореза проводят в полиакриламидном геле параллельно. Полученные ауторадиограммы позволяют сразу написать нуклеотидную последовательность, причем чтение цепи снизу вверх соответствует 5'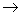 3'-полярности цепи копии. Например, положение самого короткого олигонуклеотида (в " - Т-системе") указывает на то, что следующий за ним по длине олигонуклеотид заканчивается на Т, т. е. что против полосы, расположенной выше (это полоса в - А-системе), следует записать букву Т. Таким же образом записывают далее в последовательности букву А (на основании положения следующего по длине олигонуклеотида, который оказался в "- А-системе") и т. д. Соответствующий участок цепи в матрице читается с учетом принципа комплементарности и антипараллельности цепей в комплексе матрица - затравка. Для проверки этих данных используют результаты анализа с помощью "плюс-системы". В этом случае дополнительное копирование (после первого этапа) проводят в присутствии ДНК-полимеразы, выделенной из бактериофага Т4, которая в отсутствие субстратов (нуклеозид-5'-трифосфатов) проявляет 3'-экзонуклеазную активность (аналогичную действию ФДЭ змеиного яда), т. е. отщепляет мононуклеотиды один за другим с 3'-конца. В то же время в присутствии субстратов ее полимеразная активность во много раз превосходит экзонуклеазную. Так, если в реакционной смеси присутствует хотя бы один дезоксирибонуклеозид-5'-трифосфат (dATP в " +А-системе"), деградация каждой копии, образовавшейся на первой стадии анализа, будет проходить вплоть до места положения А (Т в матрице). В этом случае pdA включается много быстрее, чем удаляется, и, таким образом, накапливаются фрагменты, содержащие на 3'-конце цепи А. Аналогично проводят копирование в присутствии только dGTP, dCTP или dTTP. Смеси параллельно подвергают электрофорезу, как и в предыдущем случае, и получают ауторадиограммы, из которых сразу считывается последовательность 5'3'-направление, считывается также снизу вверх). Из сравнения фореграмм "плюс-" и "минус-систем" делается однозначный вывод о нуклеотидной последовательности в копиях и, следовательно, в матричном полинуклеотиде.
3'-полярности цепи копии. Например, положение самого короткого олигонуклеотида (в " - Т-системе") указывает на то, что следующий за ним по длине олигонуклеотид заканчивается на Т, т. е. что против полосы, расположенной выше (это полоса в - А-системе), следует записать букву Т. Таким же образом записывают далее в последовательности букву А (на основании положения следующего по длине олигонуклеотида, который оказался в "- А-системе") и т. д. Соответствующий участок цепи в матрице читается с учетом принципа комплементарности и антипараллельности цепей в комплексе матрица - затравка. Для проверки этих данных используют результаты анализа с помощью "плюс-системы". В этом случае дополнительное копирование (после первого этапа) проводят в присутствии ДНК-полимеразы, выделенной из бактериофага Т4, которая в отсутствие субстратов (нуклеозид-5'-трифосфатов) проявляет 3'-экзонуклеазную активность (аналогичную действию ФДЭ змеиного яда), т. е. отщепляет мононуклеотиды один за другим с 3'-конца. В то же время в присутствии субстратов ее полимеразная активность во много раз превосходит экзонуклеазную. Так, если в реакционной смеси присутствует хотя бы один дезоксирибонуклеозид-5'-трифосфат (dATP в " +А-системе"), деградация каждой копии, образовавшейся на первой стадии анализа, будет проходить вплоть до места положения А (Т в матрице). В этом случае pdA включается много быстрее, чем удаляется, и, таким образом, накапливаются фрагменты, содержащие на 3'-конце цепи А. Аналогично проводят копирование в присутствии только dGTP, dCTP или dTTP. Смеси параллельно подвергают электрофорезу, как и в предыдущем случае, и получают ауторадиограммы, из которых сразу считывается последовательность 5'3'-направление, считывается также снизу вверх). Из сравнения фореграмм "плюс-" и "минус-систем" делается однозначный вывод о нуклеотидной последовательности в копиях и, следовательно, в матричном полинуклеотиде.
В настоящее время выделение фрагментов ДНК, создание рекомбинантных генов, а так же прямое секвенирование ДНК и кДНК становятся общедоступными методами благодаря широкому внедрению ПЦР (полимеразной цепной реакции).Сущность ПЦР заключается в использовании двух олигонуклеотидов-праймеров, способных специфически гибридизоваться с последовательностями нуклеотидов на противоположных концах двух цепей участка ДНК, в качестве затравки для одновременного синтеза комплементарных цепей с противоположных концов матрицы с помощью термостабильной ДНК-полимеразы. В ходе повторяющихся циклов (температурной денатурации ДНК, отжига и энзиматической достройки праймеров) экспоненциально увеличивается количество дискретного фрагмента, фланкированного последовательностями нуклеотидов, соответсвующих первичной структуре праймеров.
Применимость метода Сэнгера зависит от возможности получения одноцепочечных копий клонированных ДНК. Для этой цели можно использовать векторы на основе бактериофага М13. Двухцепочечную чужеродную ДНК можно клонировать в двухцепочечной репликативной форме (РФ) фаговой ДНК, при этом после трансформации в белковую оболочку будет упаковываться только одна из цепей ДНК. Во всех векторах типа М13тр используются сходные полилинкерные последовательности, поэтому для инициации полимеразных реакций пригоден один и тот же универсальный праймер. При амплификации смеси генов (например, семейства генов) необходимо провести клонирование ПЦР-продуктов в векторах типа М13, в результате каждый фаг будет содержать только одну вставку. При прямом секвенировании смеси генов наблюдается несколько одинаково расположенных полос в разных дорожках геля. При амплификации же одного гена можно проводить прямое секвенирование, не прибегая к промежуточному субклонированию.
Выбор оптимального праймера для ПЦР зависит от 5 '- и 3 '-концевых последовательностей амплифицируемого фрагмента ДНК. Кроме того, для встраивания ПЦР-продукта в полилинкерный сайт вектора М13 в 5'-конец праймеров должны быть включены подходящие рестрикционные сайты. В этом случае ПЦР-амплификация с последующей рестрикцией продукта позволит провести его встраивание в ДНК М13, рестрицированную тем же ферментом. В разные концы амплифицируемого фрагмента лучше включать сайты для разных рестриктаз, поскольку это позволит избежать отжига векторной ДНК самой на себя и обеспечит положение клонированной вставки в определенной ориентации (так называемое направленное клонирование). При подборе праймеров необходимо учитывать следующие факторы.
а. Следует убедиться в том, что амплифицируемое семейство генов не содержит консервативного внутреннего рестрикционного сайта, идентичного сайту, включенному в праймер.
б. После включения рестрикционного сайта 5' - конец праймера нужно удлинить, в противном случае рестриктаза не будет расщеплять праймер. Необходимая для каждого фермента длина выступающего участка и время рестрикции указаны в каталоге фирмы New England BioLabs.
Перед секвенированием двухцепочечную рекомбинантную ДНК М13 необходимо перевести в одноцепочечную форму. Для этого ее вводят путем трансформации в компетентные клетки E. сoli. Бляшки, содержащие одноцепочечные рекомбинантные фаги, необходимо выколоть, нарастить в бактериальной культуре и депротеинизировать.
Затем переносят культуру в микроцентрифужную пробирку на 1,5 мл и центрифугируют в микроцентрифуге при 12 000 g в течение 5 минут. Переносят 1 мл супернатанта (содержащего чистый фаг) во вторую пробирку на 1,5 мл, добавляют 200 мкл полиэтиленгликоля и инкубируют при комнатной температуре как минимум 15 минут. Собирают фаг центрифугированием в течении 5 минут при 12 000 g и отбирают супернатант. Быстро повторяют центрифугирование и полностью удаляют все следы супернатанта. Затем осаждают ДНК ацетатом натрия, промывают ее 70%-ным этанолом и высушивают под вакуумом. Растворяют ДНК в 30 мкл воды. Полученная ДНК представляет собой одноцепочечную матрицу для секвенирования.
Ниже приведена конкретная методика секвенирования:
Материалы
• 5 х реакционный буфер: 200 мМ трис-HCl, рН 7,5, 100 мМ MgCl2, 250 мМ NaCl
• Буфер для разведения фермента: 10 мМ трис-HCl, рН 7,5, 5 мМ ДТТ, 0,5 мг/мл БСА
• 5 х смесь длямечения: по 7,5 мкМ dGTP, dCTP, dTTP
• Смесь для ddG-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddGTP, 50 мМ NaCl
• Смесь для ddA-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddATP, 50 мМ NaCl
• Смесь для ddC-терминации: по 80 мкМ dGTP, dATP, dCTP, dTTP, 8 мкМ ddCTP, 50 мМ NaCl