Молекулярные механизмы регуляции клеточного цикла
Молекулярные механизмы регуляции клеточного цикла
Клеточный цикл тАУ это период жизни клетки от одного деления до другого или от деления до смерти. Клеточный цикл состоит из интерфазы (период вне деления) и самого клеточного деления.
В конце G1 периода принято различать специальный момент, называемый R‑точкой (точка рестрикции, R‑пункт), после которого клетка обязательно в течение нескольких часов (обычно 1тАУ2) вступает в S период. Период времени между R‑точкой и началом S периода можно рассматривать в качестве подготовительного для перехода в S период.
Самый главный процесс, который идет в S периоде, тАУ это удвоение или редупликация ДНК. Все остальные реакции, происходящие в это время в клетке, направлены на обеспечение синтеза ДНК. К таким вспомогательным процессам можно отнести синтез гистоновых белков, синтез ферментов, регулирующих и обеспечивающих синтез нуклеотидов и образование новых нитей ДНК.
Прохождение клетки по всем периодам клеточного цикла строго контролируется. При движении клеток по клеточному циклу в них появляются и исчезают, активируются и ингибируются специальные регуляторные молекулы, которые обеспечивают: 1) прохождение клетки по определенному периоду клеточного цикла и 2 переход из одного периода в другой. Причем прохождение по каждому периоду, а также переход из одного периода в другой контролируется различными веществами. Сейчас мы попробуем выяснить, что же это за вещества и что они делают.
Общая ситуация выгладит так. В клетке постоянно присутствуют специальные белки-ферменты, которые путем фосфорилирования других белков (по остаткам серина, тирозина или треонина в полипептидной цепи), регулируют активность генов, ответственных за прохождение клетки по тому или иному периоду клеточного цикла. Эти белки-ферменты называются циклин-зависимыми протеинкиназами (cdc). Имеется несколько их разновидностей, но они все обладают сходными свойствами. Хотя количество этих циклин-зависимых протеинкиназ может варьировать в различных периодах клеточного цикла, они присутствуют в клетке постоянно, независимо от периода клеточного цикла, то есть они имеются в избытке. Другими словами, их синтез или количество не лимитирует или не регулирует прохождение клеток по клеточному циклу. Однако при патологии, если синтез их нарушен, снижено их количество или имеются мутантные формы с измененными свойствами, то это, конечно же, может повлиять на течение клеточного цикла.
Почему же такие циклин-зависимые протеинкиназы сами не могут регулировать прохождение клеток по периодам клеточного цикла. Оказывается, что они находятся в клетках в неактивном состоянии, а для того чтобы они активировались и начали работать, необходимы специальные активаторы. Ими являются циклины. Их также много разных типов, но они присутствуют в клетках не постоянно: то появляются, то исчезают. В разные фазы клеточного цикла образуются разные циклины, которые связываясь с Cdk образуют различные Cdk‑циклиновые комплексы. Эти комплексы регулируют разные фазы клеточного цикла и поэтому называются G1-, G1/S-, S- и М-Cdk (рис. из моих рис. циклины). Так, например, прохождение клетки по G1 периоду клеточного цикла обеспечивает комплекс циклин-зависимой протеинкиназы‑2 (cdk2) и циклина D1, циклин-зависимой протеинкиназы‑5 (cdk5) и циклина D3. Прохождение через специальную точку рестрикции (R‑пункт) периода G1 контролирует комплекс cdc2 и циклина С. Переход клетки из G1 периода клеточного цикла в S период контролирует комплекс cdk2 и циклина Е. Для перехода клетки из S периода в G2 период необходим комплекс cdk2 и циклин А. Циклин-зависимая протеинкиназа‑2 (cdc2) и циклин В участвуют в переходе клетки из G2 периода в митоз (М период). Циклин H в соединении с cdk7 необходим для фосфорилирования и активации cdc2 в комплексе с циклином В.
| РЕГУЛЯЦИЯ КЛЕТОЧНОГО ЦИКЛА |
| G1 период | cdk2 + циклин D1
cdk5 и циклин D3 |
| R‑пункт периода G1 | cdc2 + циклин С |
| переход из G1 в S период | cdk2 + цикли Е |
| переход из S в G2 период | cdk2 + циклин А |
| переход из G2 периода в митоз (М период) | cdc2 + циклин В |
| циклин H + cdk7 необходим для фосфорилирования и активациии cdc2 в комплексе с циклином В |
Циклины тАУ новый класс белков, открытый Тимом Хантом, которые играют ключевую роль в управлении делением клеток. Название ВлциклиныВ» появилось из-за того, что концентрация белков этого класса изменяется периодически в соответствии со стадиями клеточного цикла (например, падает перед началом деления клетки).
Первый циклин был обнаружен Хантом в начале 1980-х годов, во время опытов с икрой лягушек и морских ежей. Позднее циклины были найдены и в других живых существах.
Оказалось, что эти белки мало изменились в ходе эволюции, как и механизм управления клеточным циклом, который дошел от простых дрожжевых клеток до человека в ВлзаконсервированномВ» виде.
Тимоти Хант (R. Timothy Hunt) вместе с соотечественником-англичанином Полом Нерсом (Paul M. Nurse) и американцем Лиландом Хартуэллом (Leland H. Hartwell) в 2001 году получили нобелевскую премию по физиологии и медицине за открытие генетических и молекулярных механизмов регуляции клеточного цикла тАУ процесса, который имеет важнейшее значение для роста, развития и самого существования живых организмов
Контрольные точки клеточного цикла
1. Точка выхода из G1‑фазы, называемая Старт тАУ у млекопитающих и точкой рестрикции у дрожжей. После перехода через точку рестрикции R в конце G1 наступление S становится необратимым, т.е. запускаются процессы ведущие к следующему делению клетки.
2. Точка S тАУ проверка точности репликации.
3. Точка G2/M‑перехода тАУ проверка завершения репликации.
4. Переход от метафазы к анафазе митоза.
Регуляция репликации
Перед началом репликации Sc ORC‑комплекс (origin recognition complex) садится на ori тАУ точку начала репликации. Cdc6 представлен во всем клеточном цикле, но его концентрация возрастает вначале G1, где он связывается c ОRC комплексом, к которому затем присоединяются Mcm белки с образованием pre-replicative complex (pre-RC). После сборки pre-RC клетка готова к репликации.
Для инициации репликации S-Cdk соединяется с протеинкиназой (?), которая фосфорилирует pre-RC. При этом Cdc6 диссоциирует от ОRC после начала репликации и фосфорилируется, после чего убиквитинируется SCF и деградирует. Изменения в pre-RC препятствуют повторному запуску репликации. S-Cdk так же фосфорилирует некоторые Mcm белковые комплексы, что запускает их экспорт из ядра. Последующая дефосфориляция белков вновь запустит процесс образования pre-RC.
Циклины тАУ активаторы Cdk. Циклины, так же как и Cdk вовлечены в различные, помимо контроля клеточного цикла, процессы. Циклины разделяются на 4 класса в зависимости от времени действия в клеточном цикле: G1/S, S, M и G1 циклины.
G1/S циклины (Cln1 и Cln2 у S. cerevisiae, циклин E у позвоночных) достигает максимальной концентрации в поздней G1‑фазе и падает в S‑фазе.
G1/S cyclinтАУCdk комплекс запускает начало репликации ДНК выключая различные системы подавляющие S-phase Cdk в G1‑фазе G1/S циклины также инициируют дупликацию центросом у позвоночных, образование веретенного тела у дрожжей. Падение уровня G1/S сопровождается увеличением концентрации S циклинов (Clb5, Clb6 у Sc и циклин A у позвоночных), который образует S циклин-Cdk комплекс который напрямую стимулирует ДНК репликацию. Уровень S циклина остается высоким в течении всей S, G2‑фаз и начала митоза, где помогает началу митозу в некоторых клетках.
М-циклины (Clb1,2,3 и 4 у Sc, циклин B у позвоночных) появляется последним. Его концентрация увеличивается, когда клетка переходит к митозу и достигает максимума в метафазе. М-циклин-Cdk‑комплекс включает сборку веретена деления и выравнивание сестринских хроматид. Его разрушение в анафазе приводит к выходу из митоза и цитокиезу. G1 циклины (Cln3 у Sc и циклин D у позвоночных) помогает координировать клеточный рост с входом в новый клеточный цикл. Они необычны, так как их концентрация не меняется от фазы клеточного цикла, а меняется в ответ на внешние регуляторные сигналы роста.
Программируемая клеточная гибель
В 1972 г. Керр с соавт. опубликовали статью, в которой авторы представили морфологические доказательства существования отличающегося от некроза особого вида гибели клеток, которую они назвали ВлапоптозВ». Авторы сообщили, что структурные изменения клеток при апоптозе проходят две стадии:
1-я тАУ образование апоптозных тел,
2-я тАУ их фагоцитоз и разрушение другими клетками.
Причины гибели, процессы морфологического и биохимического характера развития клеточной смерти могут быть различными. Но все же их можно четко разделить на две категории:
1. Некроз (от греч. пеkrosis тАУ омертвление) и
2. Апоптоз (от греч. корней, означающих ВлотпадениеВ» или ВлраспадениеВ»), который часто называют программируемой клеточной смертью (ПКС) или даже клеточным самоубийством (рис. 354).
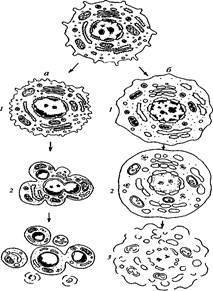
Два пути клеточной гибели
а тАУ апоптоз (профаммированная клеточная смерть): / тАУ специфическое сжатие клетки и конденсация хроматина, 2 тАУ фрагментация ядра, 3 тАУ фрагментация тела клетки на ряд апоптических телец; б тАУ некроз: / тАУ набухание клетки, вакуолярных компонентов, конденсация хроматина (кариорексис), 2 тАУ дальнейшее набухание мембранных органоидов, лизис хроматина ядра (кариолизис), 3 тАУ разрыв мембранных компонентов клетки тАУ лизис клетки
НЕКРОЗ
Н. является наиболее частой неспецефической формой гибели клеток. Он может быть вызван тяжелыми повреждениями клетки в результате прямой травмы, радиации, влияния токсических агентов, вследствие гипоксии, лизиса клетки, опосредованного комплементом и т.д.
Некротический процесс проходит ряд стадий:
1) паранекроз тАУ подобные некротическим, но обратимые изменения;
2) некробиоз тАУ необратимые дистрофические изменения, характеризующиеся преобладанием катаболических реакций над анаболическими;
3) смерть клетки, время наступления которой установить трудно;
4) аутолиз тАУ разложение мертвого субстрата под действием гидролитических ферментов погибших клеток и макрофагов. В морфологическом выражении некроз равнозначен аутолизу.
АПОПТОЗ.
Несмотря на огромное количество работ, согласованного и точного определения понятия ВлапоптозВ» нет.
Алоптоз обычно характеризовали как особую форму гибели клетки, отличную от некроза по морфологическим, биохимическим, молекулярно-генетическим и другим признакам.
А. тАУ это гибель клетки, вызываемая внутренними или внешними сигналами, которые сами по себе не являются токсичными или деструктивными. А. тАУ это активный процесс, требующий затрат энергии, транскрипции генов и синтеза белка de novo.
Обнаружено значительное количество агентов, вызывающих апоптоз этих клеток, помимо облучения и глюкокортикоидов:
- ионофоры Са2+
- аденозин
- циклический АМФ
- трибутилтин
- АТФ
- гипертермия
Изучение кинетики деградации ДНК в лимфоидных клетках in vivo и in vitro показало:
- первые отчетливые признаки распада появляются, как правило, спустя более 1 ч после воздействия, чаще к концу 2‑го часа.
- Межнуклеосомная фрагментация продолжается в течение нескольких часов и заканчивается в основном через 6, реже 12 ч после воздействия.
Сразу же от момента появления деградации при анализе обнаруживается большое количество мелких фрагментов ДНК, причем соотношение между крупными и мелкими фрагментами в ходе апоптоза значительно не меняется.
Применение ингибиторов синтеза АТФ, белка и транскрипции генов замедляет процесс апоптоза. Такой зависимости в случае Н. нет
Как видно из сравнения определений некроза и апоптоза, между двумя видами гибели клетки имеется как сходство, так и существенные различия.
Характеристика | Некроз | Апоптоз |
функционально | необратимым прекращением ее жизнедеятельности; | необратимым прекращением ее жизнедеятельности; |
морфологически | нарушением целостности мембран, изменением ядра (пикноз, рексис, лизис), цитоплазмы (отек), разрушением клетки; | потерей микроворсинок и межклеточных контактов, конденсацией хроматина и цитоплазмы, уменьшением объема клетки (сморщиванием), образованием пузырьков из плазматической мембраны, фрагментацией клетки и образованием апоптозных телец; |
биохимически | нарушением выработки энергии, коагуляцией, гидролитическим расщеплением белков, нуклеиновых кислот, липидов; | гидролизом белков цитоплазмы и межнуклеосомным распадом ДНК; |
генетически | тАУ потерей генетической информации; и завершающейся ее аутолизом или гетеролизом с воспалительной реакцией. | структурно-функциональной перестройкой генетического аппарата и завершающийся ее поглощением макрофагами и(или) другими клетками без воспалительной реакции. |
Клеточная смерть регулируется межклеточными взаимодействиями различным образом. Множество клеток многоклеточного организма нуждается в сигналах с тем, чтобы оставаться живыми. В отсутствие таких сигналов или трофических факторов в клетках развивается программа ВлсамоубийстваВ» или программируемой смерти. Например, клетки культуры нейронов погибают при отсутствии фактора роста нейронов (NGF), клетки простаты гибнут в отсутствие андрогенов семенника, клетки молочной железы тАУ при падении уровня гормона прогестерона и т.д. В то же время клетки могут получать сигналы, которые в клетках-мишенях запускают процессы, приводящие к гибели по типу апоптоза. Так, гидрокортизон вызывает гибель лимфоцитов, а глютамат тАУ нервных клеток в культуре ткани, фактор некроза опухоли (TNF) вызывает гибель самых различных клеток. Тироксин (гормон щитовидной железы) вызывает апоптоз клеток хвоста головастиков. Кроме этого существуют ситуации, когда апоптическая гибель клетки вызывается внешними факторами, например радиацией.
Понятие ВлапоптозВ» было введено при изучении гибели части клеток печени при неполной перевязке портальной вены. При этом наблюдается своеобразная картина клеточной смерти, которая затрагивает лишь отдельные клетки в паренхиме печени.
Процесс начинается с того, что соседние клетки теряют контакты, они как бы сморщиваются (первоначальное название этой формы гибели shrinkage necrosis тАУ некроз сжатием клетки), в ядрах по их периферии происходит специфическая конденсация хроматина, затем ядро фрагментируется на отдельные части, вслед за этим сама клетка фрагментируется на отдельные тельца, отграниченные плазматической мембраной, тАУ апоптические тельца.
Апоптоз тАУ процесс, приводящий не к лизису, не к растворению клетки, а к ее фрагментации, распаду. Судьба апоптических телец тоже необычна: они фагоцитируются макрофагами или даже нормальными соседними клетками. При этом не развивается воспалительная реакция.
Важно отметить, что во всех случаях апоптоза тАУ во время ли эмбрионального развития, во взрослом ли организме, в норме или при патологических процессах тАУ морфология процесса гибели клеток очень сходна. Это может говорить об общности процессов апоптоза в разных организмах и в разных органах.
Исследования на разных объектах показали, что апоптоз есть результат реализации генетически запрограммированной клеточной гибели. Первые доказательства наличия генетической программы клеточной смерти (ПКС) были получены при изучении развития нематоды Caenorhabditis elegans. Этот червь развивается всего за трое суток, и его малые размеры позволяют проследить за судьбой всех его клеток, начиная с ранних этапов дробления до половозрелого организма.
Оказалось, что при развитии Caenorhabditis elegans образуется всего 1090 клеток, из которых часть нервных клеток в количестве 131 штуки спонтанно погибает путем апоптоза и в организме остается 959 клеток. Были обнаружены мутанты, у которых процесс элиминации 131 клетки был нарушен. Были выявлены два гена сеd‑3 и сеd‑4, продукты которых вызывают апоптоз 131 клетки. Если у мутантных Caenorhabditis elegans эти гены отсутствуют или изменены, то апоптоз не наступает и взрослый организм состоит из 1090 клеток. Был найден и другой ген тАУ сеd‑9, который является супрессором апоптоза: при мутации сеd‑9 все 1090 клеток погибают. Аналог этого гена был обнаружен у человека: ген bcl‑2 также является супрессором апоптоза различных клеток. Оказалось, что оба белка, кодируемые этими генами, тАУ Сеd‑9 и Вс1тАУ2, имеют один трансмембранный домен и локализуются во внешней мембране митохондрий, ядер и эндоплазматического ретикулума.
Система развития апоптоза оказалась очень сходной у нематоды и позвоночных животных, она состоит из трех звеньев: регулятора, адаптера и эффектора. У Caenorhabditis elegans регулятором является Сеd‑9, который блокирует адаптерный белок Сеd‑4, который в свою очередь не активирует эффекторный белок Сеd‑3, протеазу, которая действует на белки цитоскелета и ядра (табл. 16).
Табл. 16. Развитие программируемой клеточной смерти (апоптоза)
| Объект | Регулятор | Адаптер | Эффектор | Результат | Результат |
| Caenorhabditis elegans | Ced‑9 ──┤ | Ced‑4 ─→ | Ced‑3 ─→ | ПКС | |
| Позвоночные | Bcl‑2 ──┤ | Apaf‑1 ─→ | Casp 9 ─→ | Casp 3 ─→ | ПКС |
Знак ──┤ тАУ торможение процессатАЪ знак ─→ тАУ стимуляцию процесса
У позвоночных система ПКС более сложная. Здесь регулятором является белок Вс1тАУ2, который ингибирует адаптерный белок Apaf‑1, стимулирующий каскад активации специальных протеиназ тАУ каспаз.
Ферменты тАУ участники процесса апоптоза
Таким образом,
- раз начавшись в клетке, такая деградация быстро протекает Влдо концаВ»;
- в апоптоз вступают не все клетки сразу или в короткий промежуток времени, а постепенно;
- разрывы ДНК происходят по линкерной (межнуклеосомной) ДНК;
- деградацию осуществляют эндо-, но не экзонуклеазы, и эти эндонуклеазы активируются или получают доступ к ДНК не в результате непосредственного взаимодействия с агентом, вызывающим апоптоз, а опосредованно, так как от момента контакта клеток с таким агентом до начала развития деградации проходит довольно значительное время, и, следовательно, фрагментация ДНК не является первой характерной ВлапоптотическойВ» реакцией клетки на молекулярном уровне. В самом деле, если бы деградация запускалась в результате непосредственного взаимодействия эндонуклеаз или хроматина с агентом, то в случае, например, действия ионизирующей радиации апоптоз происходил бы быстро и одновременно почти во всех клетках.
- Исходя из этих заключений, расшифровка молекулярного механизма развития апоптоза ВлсосредоточиласьВ» на идентификации эндонуклеаз(ы), осуществляющих фрагментацию ДНК, и механизмов, активирующих эндонуклеазы.
Эндонуклеазы
1. Деградацию осуществляет ДНКаза I. Процесс активируется Са2+ и Мg2+ [1979] и подавляется Zn2+ [1984].
Однако имеются факты, которые свидетельствуют против участия ДНКазы I в процессе фрагментации ДНК. Так известно, что этот фермент отсутствует в ядре, правда, этот аргумент не очень весомый, так как относительно небольшой размер его молекул, 31 кДа, в случае нарушения проницаемости ядерной мембраны делает участие ДНКазы I в деградации ДНК вполне реальным. Другое дело, что при обработке хроматина in vitro ДНКаза I вызывает разрывы не только в линкерной части, но и в нуклеосомной ДНК.
2. Другой эндонуклеазой, рассматриваемой в качестве основного фермента деградации ДНК, является эндонуклеаза II [Барри 1993]. Эта нуклеаза при обработке ядер и хроматина осуществляет межнуклеосомную фрагментацию ДНК. Несмотря на то, что его активность не зависит от ионов двухвалентных металлов, вопрос об участии эндонуклеазы II в деградации ДНК не снят до сих пор, поскольку фермент не только находится в лизосомах, но и выделяется из ядер клеток.
3. эндонуклеаза с молекулярной массой 18 кДа. Этот фермент был выделен из ядер погибающих путем апоптоза тимоцитов крыс [Гайдо, 1991]. Она отсутствовала в нормальных тимоцитах. Активность фермента проявляется в нейтральной среде и зависит от Са2+ и Мg2+.
4. γ-нуклеаза с молекулярной массой 31 кДа, имеющая ВлклассическуюВ» зависимость от ионов Са, Мg и Zn. Активность этого фермента повышалась в ядрах тимоцитов крыс, обработанных глюкокортикоидами [1994].
5. эндонуклеаза с молекулярной массой 22,7 кДа фермент, активность которого проявляется в ядрах тимоцитов крыс только после действия глюкокортикоидов и подавляется теми же ингибиторами, что и межнуклеосомная деградация ДНК [1993].
Каспазы
Каспазы тАУ цистеиновые протеазы, которые расщепляют белки по аспарагиновой кислоте. В клетке каспазы синтезируются в форме латентных предшественников тАУ прокаспаз. Существуют инициирующие и эффекторные каспазы. Инициирующие каспазы активируют латентные формы эффекторных каспаз. Субстратами для действия активированных каспаз служат более 60 различных белков. Это, например, киназа фокальных адгезионных структур, инактивация которой приводит к отделению апоптических клеток от соседей; это ламины, которые при действии каспаз разбираются; это цитоскелетные белки (промежуточные филаменты, актин, гельзолин), инактивация которых приводит к изменению формы клетки и к появлению на ее поверхности пузырей, которые дают начало апоптическим тельцам; это активируемая протеаза САD, которая расщепляет ДНК на олигонуклеотидные нуклео-сомные фрагменты; это ферменты репарации ДНК, подавление которых предотвращает восстановление структуры ДНК, и многие другие.
Одним из примеров разворачивания апоптозного ответа может являться реакция клетки на отсутствие сигнала от необходимого трофического фактора, например фактора роста нервов (NGF), или андрогена.
В цитоплазме клеток в присутствии трофических факторов находится в неактивной форме еще один участник реакции тАУ фосфорилированный белок Ваd. В отсутствие трофического фактора этот белок дефосфорилируется и связывается с белком Вс1тАУ2 на внешней митохондриальной мембране и этим ингибирует его антиапоптозные свойства. После этого активируется мембранный проапоптический белок Вах, открывая путь ионам, входящим в митохондрию. В это же время из митохондрий через образовавшиеся в мембране поры в цитоплазму выходит цитохром с, который связывается с адаптерным белком Араf‑1, который в свою очередь активирует прокаспазу 9. Активированная каспаза 9 запускает каскад других прокаспаз, в том числе каспазу 3, которые, будучи протеиназами, начинают переваривать мешенные белки (ламины, белки цитоскелета и др.), что вызывает апоптическую смерть клетки, ее распад на части, на апоптические тельца.
Апоптические тельца, окруженные плазматической мембраной разрушенной клетки, привлекают отдельные макрофаги, которые их поглощают и переваривают с помощью своих лизосом. Макрофаги не реагируют на соседние нормальные клетки, но узнают апоптические. Это связано с тем, что при апоптозе нарушается асимметрия плазматической мембраны и на ее поверхности появляется фосфатидилсерин, негативно заряженный фосфолипид, который в норме располагается в цитозольной части билипидной плазматической мембраны. Таким образом, путем избирательного фагоцитоза ткани как бы очищаются от погибших апоптозных клеток.
Как указывалось выше, апоптоз может быть вызван целым рядом внешних факторов, таких как радиация, действие некоторых токсинов, ингибиторов клеточного метаболизма. Необратимые повреждения ДНК вызывают апоптоз. Это связано с тем, что накапливающийся транскрипционный фактор тАУ белок р53, не только активирует белок р21, который ингибирует зависящую от циклина киназу и останавливает клеточный цикл в G1- или G2‑фазе, но и активирует экспрессию гена bax, продукт которого запускает апоптоз.
Наличие контрольных точек в клеточном цикле необходимо для определения завершения его каждой фазы. Остановка клеточного цикла происходит при повреждении ДНК в G1 периоде, при неполной репликации ДНК в S‑фазе, при повреждении ДНК в G2‑периоде и при нарушении связи веретена деления с хромосомами.
Одним из контрольных пунктов в клеточном цикле является собственно митоз, который не переходит в анафазу при неправильной сборке веретена и при отсутствии полных связей микротрубочек с кинетохорами. В этом случае не происходит активации АРС-комплекса, не происходит деградации когезинов, соединяющих сестринские хрома-тиды, и деградации митотических циклинов, что необходимо для перехода в анафазу.
Повреждения ДНК препятствуют вхождению клеток в S‑период или в митоз. Если эти повреждения не катастрофические и могут быть восстановлены за счет репаративного синтеза ДНК, то блок клеточного цикла снимается, и цикл доходит до своего завершения. Если же повреждения ДНК значительные, то каким-то образом происходят стабилизация и накопление белка р53, концентрация которого в норме очень низкая из-за его нестабильности. Белок р53 является одним из факторов транскрипции, который стимулирует синтез белка р21, являющегося ингибитором комплекса СDК-циклин. Это приводит к тому, что клеточный цикл останавливается на стадии G1или G2. При блоке в G1‑периоде клетка с повреждением ДНК не вступает в S‑фазу, так как это могло бы привести к появлению мутантных клеток, среди которых могут быть и опухолевые клетки. Блокада в G2‑периоде также предотвращает процесс митоза клеток с повреждениями ДНК. Такие клетки, с блокированным клеточным циклом, в дальнейшем погибают путем апоптоза, программированной клеточной гибели (рис. 353).
При мутациях, приводящих к потере генов белка р53, или при их изменениях, блокады клеточного цикла не происходит, клетки вступают в митоз, что приводит к появлению мутантных клеток, большая часть из которых нежизнеспособна, другая тАУ дает начало злокачественным клеткам.
Избирательные повреждения митохондрий, при которых в цитоплазму высвобождается цитохром с, также являются частой причиной развития апоптоза. Особенно митохондрии и другие клеточные компоненты страдают при образовании токсически активных форм кислорода (АТК), под действием которых во внутренней мембране митохондрий образуются неспецифические каналы с высокой проницаемостью для ионов, в результате чего матрикс митохондрий набухает, а внешняя мембрана разрывается. При этом растворенные в межмембранном пространстве белки вместе с цитохромом с выходят в цитоплазму. Среди освободившихся белков есть факторы, активирующие апоптоз, и прокаспаза 9.
Многие токсины (рицин, дифтерийный токсин и др.), а также антиметаболиты могут вызывать гибель клеток путем апоптоза. При нарушении синтеза белка в эндоплазматическом ретикулуме в развитии апоптоза участвует локализованная там прокаспаза 12, которая активирует ряд других каспаз, и в том числе каспазу 3.
Элиминация тАУ удаление отдельных клеток путем апоптоза, наблюдается и у растений. Здесь апоптоз включает в себя, так же как у животных клеток, фазу индукции, эффекторную фазу и фазу деградации. Морфология гибели клеток растений сходна с изменениями клеток животных: конденсация хроматина и фрагментация ядра, олигонуклеотидная деградация ДНК, сжатие протопласта, его дробление на везикулы, разрыв плазмодесм и т.д. Однако везикулы протопласта разрушаются гидролазами самих везикул, так как у растений нет клеток, аналогичных фагоцитам. Так, ПКС происходит при росте клеток корневого чехлика, при формировании перфораций у листьев, при образовании ксилемы и флоэмы. Опадание листьев связано с избирательной гибелью клеток определенной зоны черенка.
Биологическая роль апоптоза, или программированной смерти клеток, очень велика: это удаление отработавших свое или ненужных на данном этапе развития клеток, а также удаление измененных или патологических клеток, особенно мутантных или зараженных вирусами.
Итак, для того чтобы клетки в многоклеточном организме существовали, нужны сигналы на их выживание тАУ трофические факторы, сигнальные молекулы. Эти сигналы могут быть переданы на расстояние и уловлены соответствующими рецепторными молекулами на клетках-мишенях (гормональная, эндокринная сигнализация), это может быть паракринная связь, когда сигнал передается на соседнюю клетку (например, передача нейромедиатора). При отсутствии таких трофических факторов реализуется программа апоптоза. В то же время апоптоз может вызываться сигнальными молекулами, например при резорбции хвоста головастиков под действием тироксина. Кроме того, действие ряда токсинов, влияющих на отдельные звенья метаболизма клетки, также может стать причиной клеточной гибели посредством апоптоза.
Апоптоз в патогенеза заболеваний
1. В иммунной системе
2. ОНКОЛОГИЧЕСКИЕ ЗАБОЛЕВАНИЯ
3. ВИРУСНАЯ ИНФЕКЦИЯ (индуцирующие апоптоз: в. иммунодефицита человекатАЪ в. анемии циплят; ингибирующие апоптоз: цитомегаловирустАЪ в. Эпштейна-БарртАЪ в. герпеса)
4. А. и НЕЙРОНЫ КОРЫ ГОЛОВНОГО МОЗГА
ПРИНЦИПЫ КОРРЕКЦИИ АПОПТОЗА КЛЕТКИ
Открытие регулируемого процесса гибели клетки тАУ апоптозатАУпозволило определенным образом воздействовать на его отдельные этапы с целью регуляции или коррекции.
Биохимические процессы развития апоптоза можно гипотетически разделить на несколько этапов:
- действие фактора, вызывающего апоптоз;
- передача сигнала с рецепторной молекулы в клеточное ядро;
- активация апоптозспецифических генов;
- синтез апоптозспецифических белков
- активация эндонуклеаз
- фрагментацию ДНК (рис. 2.4).
В настоящее время считают, что если клетка погибает путем апоптоза, то подразумевается возможность терапевтического вмешательства, если вследствие некроза, то такое вмешательство невозможно. На основе знаний регуляции запрограммированной гибели клетки используется широкий ряд препаратов с целью воздействия на этот процесс в различных типах клеток.
Так, сведения о рецепторопосредованной регуляции апоптоза клеток учитывают при лечении гормонзависимых опухолей.
- Андрогенблокирующую терапию назначают при раке предстательной железы.
- Рак молочной железы часто подвергается регрессии при использовании антагонистов эстрогеновых рецепторов.
- Информация о биохимических сигналпередающих путях регуляции апоптоза позволяет эффективно применять антиоксидантную терапию, препараты, регулирующие концентрацию кальция, активаторы или ингибиторы различных протеинкиназ и т.д. с целью коррекции апоптоза в различных типах клеток.
Осознание роли апоптоза в гибели клеток интенсифицировало поиск фармакологических воздействий, защищающих клетки от апоптоза.
- Активно изучаются ингибиторы специфических протеаз в качестве фармакологических агентов. Это, как правило, три- или тетрапептиды, содержащие аспарагиновую кислоту (Асп). Использование таких протеаз в терапевтических целях ограничено их низкой способностью проникать в клетку. Однако, несмотря на это, в экспериментах in vivo успешно применяется Z-VAD-FMK [N‑бензилоксикарбонил-Вал-Ала-Асп(ОМе) тАУ фторметилкетон] тАУ ингибитор ICE‑подобных протеаз широкого спектра действия для снижения зоны инфаркта при моделировании инсульта.
- В ближайшие годы можно ожидать появления новых лекарственных средств для лечения и предупреждения различных заболеваний, основу действия которых будет составлять принцип регуляции процессов апоптоза.
Наиболее эффективны для коррекции апоптоза подходы, связанные с регуляцией апоптозспецифических генов. Эти подходы лежат в основе генной терапии тАУ одного из перспективных направлений лечения больных с заболеваниями, вызванными нарушением функционирования отдельных генов.
Принципы генной терапии включают следующие этапы:
тАв идентификация последовательности ДНК, которая будет подвергаться лечению;
тАв определение типа клеток, в которых будет проводиться лечение;
тАв защита ДНК от гидролиза эндонуклеазами;
тАв транспорт ДНК в клетку (ядро).
Геннотерапевтические подходы позволяют
- усиливать работу отдельных генов (трансформация генов, ингибирующих апоптоз, например гена bcl‑2),
- ослаблять их экспрессию. Для селективного ингибирования экспрессии генов в настоящее время используют технику антисмысловых олигонуклеотидов (антисенсов). Использование антисенсов снижает синтез определенных белков, что влияет на регуляцию процесса апоптоза.
Механизм действия антисенсов активно изучается. В некоторых случаях короткие (13тАУ17 оснований) антисмысловые олигонуклеотиды, имеющие последовательности, комплементарные нуклеотидным последовательностям матричной РНК (мРНК) отдельных белков, могут эффективно блокировать генетическую информацию на стадии, предшествующей транскрипции (рис. 2.5). Данные олигонуклеотиды, связываясь с ДНК, формируют триплетную спиральную структуру. Такое связывание может быть необратимым или вызывать селективное выщепление триплетного комплекса, что в итоге приводит к ингибированию экспрессии гена и гибели клетки. В других случаях происходит комплементарное связывание антисенса с мРНК, что вызывает нарушение трансляции и снижение концентрации соответствующего белка.
Триплетный комплекс

Рис. Регуляция экспрессии генов антисмысловыми олигонуклеотидами.
В настоящее время убедительно показано, что технология с использованием антисенсов имеет большое значение для регуляции отдельных генов в культуре клеток. Успешное подавление гена bcl‑2 в экспериментах на культурах клеток пробуждает надежду на применение в будущем антисенсов для лечения больных раком. Во многих экспериментах in vitro показано, что антисенсы вызывают ингибирование пролиферации и дифференцировки клеток. Такой результат подтверждает перспективы терапевтического использования данной технологии.
Вместе с этим смотрят:
G-белки и их функция
Австралопитеки - обезьянолюди или человекообезьяны?
Адаптация микроорганизмов в экстремальных условиях космоса
Адвентивна флора Чернiгiвськоi областi: iсторiя формування та сучасний стан
Адсорбция ионных и неионных поверхностно-активных веществ (ПАВ)