Обнаружение единичных нуклеотидных замен в ДНК: расщепление РНКазой и денатурирующий градиентный гель-электрофорез
Методы обнаружения нуклеотидных замен в геномной ДНК позволили исследователям разобраться в природе многих наследственных болезней человека. Эти методы дают возможность идентифицировать специфические мутации, приводящие к заболеванию, а также полиморфные участки ДНК, используемые в качестве маркеров в генетическом анализе. Благодаря развитию методов выявления нуклеотидных замен стала реальностью пренатальная диагностика многих наследственных болезней человека. Если ген, отвечающий за заболевание, известен, соответствующую мутацию можно обнаружить в геномной ДНК или в РНК при помощи блот-гибридизации с использованием меченых олигонуклеотидов в качестве гибридизационных зондов. В том случае, когда мутировавшая нуклеотидная последовательность неизвестна, замены нуклеотидов можно определить по полиморфизму длины рестрикционных фрагментов. ПДРФ обнаруживается по наличию или отсутствию сайта рестрикции во фрагменте геномной ДНК при гибридизации меченого ДНК-зонда с обработанной рестриктазами геномной ДНК, расфракционированной по размеру в агарозном геле и перенесенной на мембранный фильтр. Этот метод оказался очень эффективным для выявления как значимых мутаций, так и нейтрального полиморфизма в геноме человека и других организмов. Однако большую часть мутаций и полиморфных участков генома не удается обнаружить с помощью анализа ПДРФ, поскольку вероятность того, что замена нуклеотида изменит именно сайт рестрикции, низка. Так, например, многие точковые мутации гена р-глобина человека, вызывающие талассемию, не изменяют сайтов рестрикции, а потому не могут быть непосредственно определены прн анализе ПДРФ. Кроме того, оказалось, что в некоторых участках генома млекопитающих полиморфизм незначителен, что крайне затрудняет выявление в них ПДРФ даже при использовании большого числа различных рестриктаз. Каждый из них позволяет идентифицировать по крайней мере 50% всех возможных замен нуклеотидов на участке ДНК Длиной до 1000 п.н.
Задача настоящей работы тАУ подробно изложить суть новых методов, обсудить их преимущества и недостатки, рассмотреть некоторые модификации. Кроме того, мы на примерах покажем, как можно использовать предложенные нами методы для исследования фрагментов ДНК, полученных при амплификации специфических последовательностей в ходе полимеразной цепной реакции.
1. Общее описание методов
1.1 РНКазное расщепление
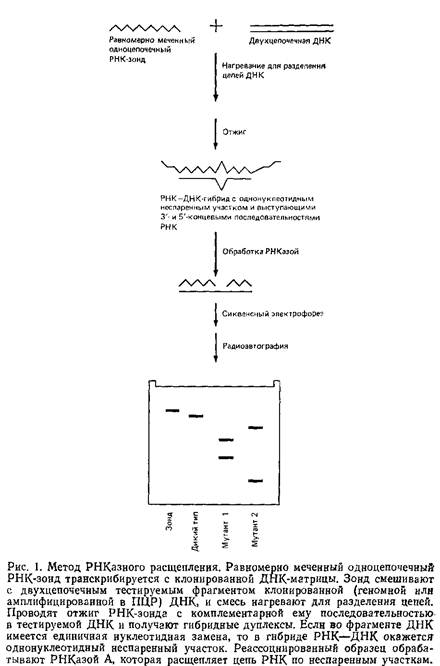
Единичные нуклеотндные замены во фрагментах геномной ДНК можно обнаружить при расщеплении неспаренных участков в РНК-ДНК-дуплексах, обрабатывая эти дуплексы РНКазой А. Рис. 1 иллюстрирует последовательные этапы процедуры.
1. Синтез равномерно меченного одноцепочечного РНК-зонда с использованием транскрипции in vitro клонированного фрагмента ДНК.
2. Гибридизация зонда с комплементарными последовательностями геномных ДНК тАУ Если в гибриднзуемой ДНК имеется замена нуклеотида, образующие РНКтАУДНК-дуплексы содержат однонуклеотндные неспаренные участки.
3. Обработка дуплекса РНКазой А, приводящая к расщеплению цепи РНК по многим, хотя и не по всем, неспаренным нуклеотидам.
4. Анализ меченых фрагментов РНК при помощи денатурирующего гель-электрофореза с последующей радиоавтографией. В этом случае эффективное расщепление по неспаренным основаниям приводит к появлению на радиоавтографах двух меченых фрагментов РНК, размеры которых указывают на положение нуклеотидной замены относительно концов фрагмента ДНК. Одиночный меченый фрагмент РНК, равный по длине фрагменту ДНК, наблюдается в том случае, если в последнем нет нуклеотидных замен или если существующие замены препятствуют расщеплению РНКтАУДНК-Дуплекса РНКазой.
На рис. 2 в качестве примера представлены результаты реакций РНКазного расщепления. При такой обработке расщепляется примерно 30тАУ40% от общего числа неспаренных участков в РНКтАУДНК-дуплексе. Тестирование фрагмента ДНК в двух независимых реакциях РНКазного расщепления с двумя зондами, комплементарными каждой из его цепей, позволяет выявить в нем 60тАУ70%: всех возможных замен нуклеотидов, так как в этом случае обнаруживаются даже комплементарные замены в обеих цепях.
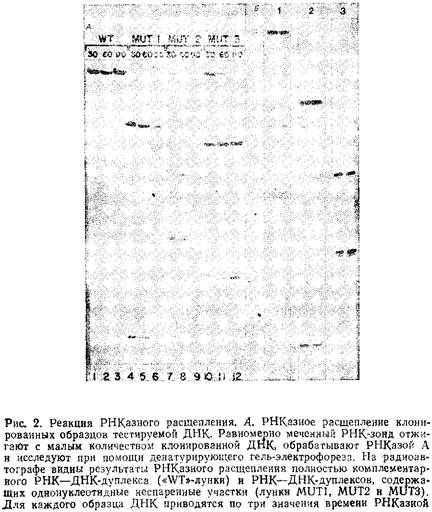
Оптимальными, на наш взгляд, являются РНК-зонды и соответственно исследуемые на однонуклеотидные замены участки ДНК длиной от 100 до 1000 нуклеотидов. В этом случае и сам зонд, и продукты его расщепления легко выявляются при помощи денатурирующего гель-электрофореза в полиакриламидном геле, аналогичном используемому при секвенировании ДНК. При этом отношение сигнала к фону достаточно высоко для получения четких результатов. Совсем нетрудно наработать меченые одноцепочечные РНК-зонды гораздо большей длины, но результаты обработки таких длинных цепей РНКазой неоднозначны. Еще одна проблема, которая возникает при использовании зондов с длиной цепи более 1000 п.и., заключается в том, что под действием РНКазы наряду с расщеплением РНКтАУДНК-дуплексов по неспаренным участкам может происходить расщепление цепи РНК по комплементарно спаренным участкам дуплекса. Кроме того, анализ продуктов расщепления длинных РНК-зондов требует проведения денатурирующего электрофореза в агарозном геле. При этом в ряде случаев не удается полностью разделить гибриды РШтАУДШ и результаты получаются противоречивыми, так как фрагменты РНК, полученные при расщеплении ее по неспаренным участкам, но оставшиеся связанными с ДНК, обнаруживаются в геле в зонах, соответствующих длине всего зонда. Для более эффективного выявления единичных нуклеотидных замен путем РНКазного расщепления мы использовали по несколько зондов в каждой реакции отжига / расщепления. Однако полученные таким способом результаты зачастую очень неоднозначны и с трудом поддаются интерпретации. В силу перечисленных причин при исследовании фрагментов ДНК на единичные замены мы бы рекомендовали использовать в реакциях РНКазного расщепления РНК-зонды длиной от 100 до 1000 нуклеотидов.
Показано, что рибонуклеаза может расщеплять неспаренные участки и в дуплексах РНКтАУРНК. По данным Перучо, единичные мутации в H-ras-гене выявляются при расщеплении неспаренных участков дуплекса РНКтАУРНК, образованного мРНК гена H-ras и его ВлантисмысловойВ» мРНК. Наши исследования показали, что естественные мутации в гене р-глобина и искусственные мутации в промоторном участке гена р-интерферона можно обнаружить при РНКазном расщеплении как РНК тАУ ДНК-, так и РНКтАУРНК-дуплексов. На клонированной ДНК-матрице мы получали равномерно меченные одноцепочечные зонды Влантисмысловой РНКВ», ренатурировали их с комплементарной мРНК, обрабатывали образовавшиеся дуплексы РНКазой, а затем идентифицировали продукты расщепления денатурирующим электрофорезом в полиакриламидном геле. Поэтому приведенные ниже методики для исследования образцов ДНК с равным успехом могут использоваться и при работе с РНК.
1.2 Денатурирующий градиентный гель-электрофорез
Два фрагмента ДНК, различающиеся лишь делецией, инсерцией или заменой одного нуклеотида, или единственным неспаренным нуклеотидом, можно легко разделить при помощи денатурирующего градиентного гель-электрофореза, ДГГЭ. Принцип метода заключается в разделении двухцепочечных фрагментов ДНК электрофорезом в стандартном акриламидном геле с линейным градиентом денатурирующих факторов тАУ мочевины, формамида или температуры. Ниже приводятся теоретические основы метода. Здесь отметим только, что разделение очень похожих фрагментов в геле происходит из-за разных температур плавления их ДНК.
1.2.1 Теоретические основы метода ДГГЭ
При постепенном повышении температуры или концентрации денатурирующего вещества происходит плавление двухцепочечной ДНК, и для каждого фрагмента параметры этого процесса различны. Внутри фрагмента имеются так называемые области плавления тАУ блоки последовательностей, плавящихся одновременно при определенных дискретных значениях температуры, которая и является температурой плавления данной области. Длина таких областей составляет от 25 до нескольких сотен нуклеотидных пар. Два соседних домена могут иметь четкую границу и различаться по Гт на несколько градусов. Фрагменты ДНК длиной 100тАУ1000 п.н. имеют обычно от двух до пяти областей плавления. Температура плавления доменов в рестрикционных фрагментах большинства образцов ДНК из природных источников колеблется от 65 до 80В°С.
Общеизвестно, что нуклеотидный состав фрагментов ДНК влияет на температуру их плавления. Оптимальные условия для отжига и отмывки нуклеиновых кислот в реакциях гибридизации подбирают исходя из их нуклеотидного состава. В гораздо меньшей степени учитывается вклад стэкинг-взаимодействий в термодинамическую стабильность двойной спирали. А между тем энергия таких взаимодействий между соседними нуклеотидами одной цепи ДНК, удерживающих ее в скрученном состоянии в физиологических растворах, больше энергии водородных связей комплементарного спаривания. Порядок чередования оснований определяет степень стэкинга и, следовательно, влияет на термостабильность фрагмента ДНК. Даже единичная нуклеотидная замена может так сильно сказаться на стэкинг-взаимодействий, что Гт изменится более чем на 1В°С. Поскольку процесс плавления домена практически полностью определяется кооперативными взаимодействиями, любая единичная нуклеотидная замена в любой его точке будет менять температуру плавления,
Разработанный Лерманом и Фишером метод электрофоретического разделения близких последовательностей ДНК основан на использовании различий в температуре плавления, обусловленных нуклеотидными заменами. Они исходят из того факта, что последовательность оснований влияет на Тт области плавления и что конформация фрагмента ДНК обусловливает его подвижность в геле под действием электрического поля. Принцип метода тАУ электрофорез ДНК в акриламидном геле фиксированной концентрации с линейно возрастающим к нижней части геля градиентом концентрации веществ, денатурирующих ДНК: обычно мочевины или формамида. Электрофорезная камера нагревается до температуры примерно 60В°С.
Это чуть ниже температуры плавления самой легкоплавкой области во фрагменте ДНК тАУ двухцепочечная молекула ДНК входит в гель и продвигается в нем со скоростью, пропорциональной ее молекулярной массе. Как только фрагмент достигает участка геля, в котором температура камеры и концентрация денатурирующих веществ создают условия для плавления первой, самой легкоплавкой области, он как бы разветвляется: одна часть его остается двухцепочечной, а другая переходит в одноцепочечную форму. Такая разветвленная структура застревает в порах геля, в результате чего ее подвижность снижается. Снижение электрофоретической подвижности молекулы коррелирует с соотношением длин денатурированного и двухцепочечного участков: чем длиннее самый легкоплавкий домен, тем сильнее снижается подвижность. Участок геля, в котором молекула ДНК начинает терять скорость, соответствует самым легкоплавким доменам. Если концентрации денатурирующих веществ подобраны правильно, то два фрагмента ДНК, различающиеся лишь одной нуклеотидной заменой и имеющие неодинаковые значения Тт., начнут снижать скорость в различных участках геля, и к концу прохождения через него их легко будет разделить. Ранее считалось, что отделить мутировавшие фрагменты от фрагмента ДНК дикого типа за счет разной степени снижения их подвижности в ДГГЭ можно лишь в том случае, если замены произошли в области с самым низким значением Тт. Однако анализ большого числа образцов ДНК, а также последующие теоретические выкладки показали, что в большинстве случаев метод ДГГЭ дает возможность обнаружить нуклеотидные замены во всех областях плавления, за исключением самых термостабильных. Замены в областях с самой высокой температурой плавления обычно не обнаруживаются, так как по окончании плавления последнего домена ДНК полностью денатурирует, а это приводит к нарушению корреляции между конформацией молекулы и скоростью ее продвижения. Важно помнить, что успех при проведении ДГГЭ с целью разделения мутировавшего фрагмента и фрагмента ДНК дикого типа определяется структурой фрагмента. Она должна быть разветвленной, причем мутации должны приходиться на области, подвергшиеся плавлению. Для увеличения разрешающей способности метода к исследуемому фрагменту ДНК ВлпришивалиВ» последовательность с более высокой температурой плавления ДНК, так называемый ВлGC-зажимВ». При этом все области плавления фрагмента, включая самую тугоплавкую, становились доступными для анализа. Эти опыты проводили с препаратами ДНК, клонированными в плазмидном векторе, содержащем ВлGC-зажимВ». Чтобы избежать стадии клонирования, фрагменты с ВлзажимамиВ» можно получить в полимеразной цепной реакции.
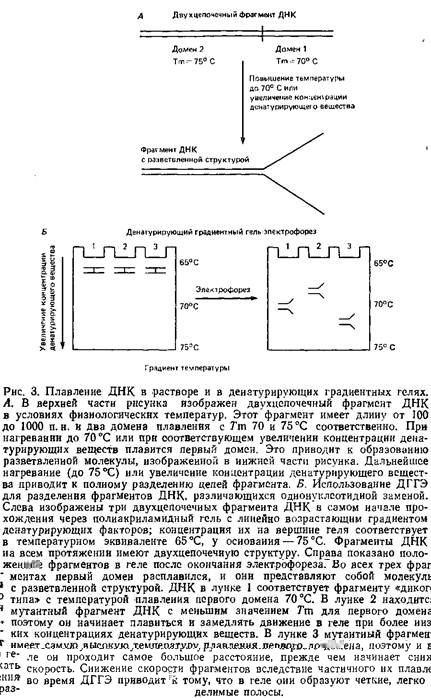
Еще одним усовершенствованием ДГГЭ следует считать использование гетеродуплексных фрагментов, образованных мутировавшей ДНК и ДНК дикого типа. В предыдущих опытах сравнивали подвижность гомодуплексных фрагментов мутантной и исходной ДНК в параллельных дорожках при ДГГЭ. Работа с полученными из них гетеродуплексами позволяет значительно повысить разрешающую способность метода и долю обнаруживаемых мутаций. Причина заключена в том, что неспаренные участки гетеродуплекса, в которых имеются однонуклеотидные замены, сильно снижают стзкинг-взаимодействия оснований, дестабилизируя вторичную структуру, а это, в свою очередь, приводит к снижению температуры плавления фрагмента ДНК- В ряде случаев замена всего одного нуклеотида обусловливала снижение температуры на целых 6В°С. Анализ большого числа гетеродуплексов и теоретические подсчеты показывают, что все возможные замены нуклеотидов в легкоплавких доменах, приводящие к образованию неспаренных участков во фрагменте ДНК, выявляются при помощи ДГГЭ с вероятностью 100% за счет изменения подвижности. На долю легкоплавких областей во фрагментах ДНК от 100 до 1000 п.н. приходится в среднем более 50% их длины, следовательно, использование гетеродуплексов таких фрагментов позволяет выявить более половины всех возможных замен нуклеотидов. В клонированных же фрагментах ДНК с ВлGC-зажимомВ» выявляются практически все замены нуклеотидов.
Гетеродуплексы дают возможность наблюдать изменение подвижности мутантных фрагментов ДНК в геле, не выявляемое при стандартных условиях проведения электрофореза. Зачастую единичные мутации можно обнаружить по вызываемым ими значительным изменениям структуры доменов во фрагменте ДНК-Изменения эти отражаются и на характере плавления ДНК: домены, имевшие изначально самую высокую температуру плавления, могут стать самыми легкоплавкими. Таким образом, когда вместо гомодуплексов используются гетеродуплексы, ДГГЭ позволяет выявлять замены нуклеотидов даже в самых тугоплавких доменах.
1.2.2 Практическое использование ДГГЭ
Приведенные здесь схемы ДГГЭ обеспечивают более высокую разрешающую способность и эффективность обнаружения мутаций за счет использования гетеродуплексов. Их основные этапы приведены на рис. 4. Исследуемые препараты ДНК отжигают с меченым одноцепочечным ДНК-зондом, и если в них произошла замена нуклеотида, образовавшийся гетеродуплекс будет иметь однонуклеотидный неспаренный участок. Далее проводят электрофорез в денатурирующем геле с последующей радиоавтографией, используя в качестве контроля гомодуплекс ДНК дикого типа. Таким образом, в этом случае не требуется стадии блотинга геля. Кроме одноцепочечных ДНК-зондов можно использовать асимметрично меченные двухцепочечные ДНК-зонды, а также меченые одноцепочечные РНК-зонды для тестирования ДНК в гибридах РНКтАУДНК или РНК в РНКтАУРНК-гибридах.
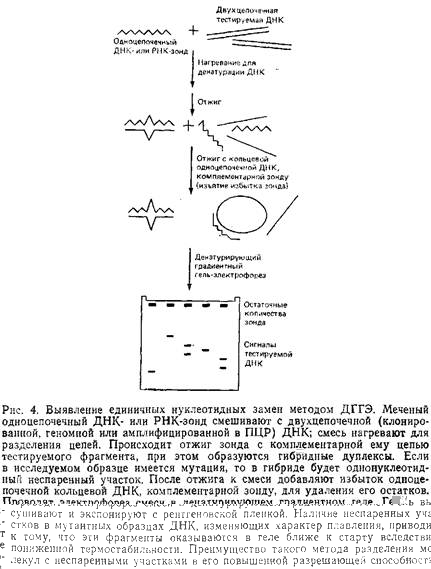
С помощью ДГГЭ, также как и при РНКазном расщеплении, можно исследовать фрагменты нуклеиновых кислот длиной от 100 до 1000 п.н. Верхний предел значений длин в некоторой степени определяется необходимостью использовать полиакриламидный гель. Подвижность в нем фрагментов ДНК длиной более 1000 п. н. резко снижается, а это значительно увеличивает время, необходимое для их электрофоретического разделения. Еще более серьезную проблему представляет собой характер плавления длинных фрагментов. Известно, что чем длиннее фрагмент, тем. больше в нем доменов плавления. При прохождении такого фрагмента через гель очень быстро наступает резкое снижение его подвижности, обусловленное плавлением одновременно большого числа доменов, что делает доступной для обнаружения замены лишь незначительную часть длинного фрагмента. В силу вышеуказанных причин мы стараемся работать с фрагментами, длина которых не превышает 1000 п. и. Поскольку для большинства фрагментов ДНК более половины их длины приходится на легкоплавкие домены, то денатурирующий электрофорез фрагмента в 1000 п. и. позволяет тестировать на нуклеотидные замены примерно 500 нуклеотидов. Для повышения информативности анализа можно использовать ДНК-зонд длиной 1000тАУ2000 п.н.; отжигать его с геномной ДНК, обрабатывать соответствующими рестриктазами и получать при этом два-три фрагмента оптимального размера.
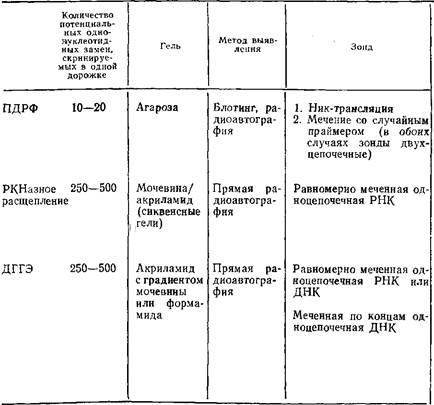
Таблица 1. Сравнительная характеристика методов градиентного анализа ПДРФ, рестриктазного расщепления и денатурирующего гель-электрофореза

1.3. Полимеразная цепная реакция
РНКазное расщепление и ДГГЭ можно использовать для непосредственного исследования фрагментов геномной ДНК, минуя стадию клонирования. Работая с равномерно меченными зондами, обладающими высокой удельной активностью, можно любым из этих методов получить желаемый результат, имея изначально 5тАУ10 мкг геномной ДНК человека и проводя радиоавтографию 24 ч. Чем проще организм, тем выше чувствительность методов. Хотя получаемые результаты в большинстве своем можно оценить высоко, непосредственное использование суммарной геномной ДНК ставит перед исследователем ряд проблем. Это, во-первых, нередко низкое отношение сигнала к фону, особенно при РНКазном расщеплении. Во-вторых, активность равномерно меченных фосфором зондов должна быть настолько высокой, чтобы использовать их в течение дня или двух. В-третьих, при проведении некоторых анализов, особенно в случае множественных тестов, лимитирующим фактором может стать количество геномной ДНК. Любой тест тем предпочтительнее, чем меньшее количество ДНК требует.
В 1985 г. был описан принципиально новый метод, позволяющий в миллион раз амплифицировать интересующие последовательности в препарате геномной ДНК, тАУ полимеразная цепная реакция, ПЦР. Идея метода и ее воплощение очень просты. Сначала синтезируются два дезоксиолигонуклеотида длиной 20тАУ30 оснований, представляющие собой концевые последовательности интересующего фрагмента ДНК. Полярность выбрана так, чтобы после отжига их направления 5'-3' были обращены друг к другу. Избыточные количества этих олигонуклеотидов смешивают с геномной ДНК, и смесь нагревают для денатурации последней. Снижение температуры приводит к реассоциации олигонуклеотидов с гомологичными участками геномной ДНК. Затем проводят наращивание цепи при участии ДНК-полимер азы и дезоксирибонуклеотидтрифосфатов. Такая последовательность реакций денатурации, реассоциации и наращивания цепи повторяется 20тАУ30 раз. Уже после двух циклов среди продуктов реакции появляются фрагменты ДНК, точно совпадающие по длине с исходным фрагментом, ограниченным олигонуклеотидами. Эти фрагменты служат матрицей для последующих реакций и идентичны большинству конечных продуктов. Процесс является по существу цепным, так как продукты данной реакции служат матрицей для последующих реакций. Количество вновь образующейся ДНК возрастает в геометрической прогрессии, поэтому за 20 циклов при 100% тАУ ной эффективности каждого из них можно получить 220 молекул. На практике эффективность каждого цикла амплификации составляет 20тАУ50%, т.е. при проведении достаточного числа циклов можно добиться увеличения количества специфической последовательности кратного миллиону.
Если при амплификации геномной ДНК позвоночных в качестве праймеров для ПЦР используют олигонуклеотиды длиной 20 нуклеотидов и более, то процесс этот довольно специфичен и амплифицируется только один фрагмент ДНК тАУ Однако иногда среди продуктов реакции наблюдается накопление фрагментов, происхождение которых трудно объяснить. А поскольку эти фрагменты могут мешать последующему анализу, то рекомендуется провести еще одну серию амплификации, с использованием другого набора олигонуклеотидных праймеров. Этот метод, именуемый Влnested oligoВ», состоит в следующем: продукт первичной полимеразной цепной реакции используется в качестве матрицы в последующих раундах ПЦР, но уже с двумя другими олигонуклеотидами, имеющими гомологии с участками ДНК внутри первичного амплифицированного фрагмента. Такая процедура позволяет получить большое количество индивидуальной последовательности ДНК, несколько более короткой, чем исходный фрагмент, и использовать ее для последующего анализа.
Имея исходно менее 1 мкг суммарной геномной ДНК позвоночных, в ПЦР можно получить несколько микрограмм специфического фрагмента. Хорошо амплифицируются фрагменты до 2000 п. н. В одной реакции можно амплифицировать одновременно и несколько фрагментов. Амплификация специфических фрагментов с помощью ПЦР может применяться и в диагностике, и при клонировании. Левинсон и Гитшейер использовали амплифицированную в ПЦР геномную РНК и РНКазное расщепление для выявления однонуклеотидных замен в гене фактора VIII, обусловливающих Х-сцепленную гемофилию А человека. Мы объясняем, как применять ПЦР в сочетании с РНКазным расщеплением и с ДГГЭ для обнаружения однонуклеотидных замен в препаратах геномной ДНК.

3. Предварительные процедуры
Для проведения ПЦР, РНКазного расщепления и ДГГЭ необходимы следующие предварительные процедуры.
1. Фрагмент ДНК, тестируемый на мутации или полиморфизм, необходимо клонировать в плазмидном векторе, позволяющем синтезировать определенные типы зондов.
2. Очень полезно, а иногда и просто необходимо иметь ре-стрикционную карту такой клонированной вставки ДНК тАУ Расположение сайтов рестрикции, особенно тех, что встречаются лишь 1тАУ2 раза во всей плазмиде, можно определить при помощи стандартных методик рестрикционного картирования. Для обоих типов одноцепочечных зондов необходимо иметь единичный сайт рестрикции в участке тестируемой ДНК, дистальном по отношению к сайту связывания с РНК-полимеразой в случае РНК-зондов или с олигонуклеотидами в случае ДНК-зондов. После реассоциации одноцепочечного меченого ДНК-зонда с тестируемой ДНК может возникнуть необходимость расщепить гибридную последовательность ДНК на 2тАУ3 фрагмента с оптимальной для ДГГЭ длиной. В этом случае надо использовать сайты рестрикции, имеющиеся в тестируемом фрагменте.
3. При использовании ПЦР необходимо определить концевые нуклеотидные последовательности тестируемой вставки ДНК. Обычно требуется секвенировать 40тАУ50 п. н., чтобы получить первый набор олигонуклеотидов длиной 20тАУ25 п. н. При работе методом Влnested oligoВ» необходимо дополнительно просеквенировать любую последовательность внутри фрагмента ДНК, ограниченного первой парой олигонуклеотидов.
4. Метод РНКазного расщепления
Основное внимание уделено экспериментальным особенностям каждой стадии процесса РНКазного расщепления, включая получение меченого РНК-зонда. Большинство этапов аналогично описываемым в оригинальных работах, но приведены и некоторые модификации. Обсуждаются также возможные методические трудности и способы их преодоления.
Отметим, что для данного метода необязательно, чтобы концевые участки РНК-зонда и тестируемого образца ДНК были строго комплементарны. Выступающие некомплементарные 5'или З'-концы зонда не мешают анализу, так как они просто разрушаются РНКазой в процессе реакции расщепления. В принципе даже полезно иметь выступающие концы, удлиняющие зонд на 5тАУ10% по сравнению с фрагментом ДНК тАУ Это позволяет различать зонд и гибридный фрагмент дикого типа, получающийся при РНКазном расщеплении. Кроме того, выступающие концы дают возможность проверить активность РНКазы: если фермент инактивирован или активность его недостаточна, то не происходит их эффективного удаления и они обнаруживаются при радиоавтографии.
4.1 Материалы
4.1.1 Реактивы для получения РНК-зондов
Для получения равномерно меченных РНК-зондов необходимы те же реактивы, что и для реакций РНКазного расщепления. Здесь мы приводим условия использования РНК-полиме-раз и промоторов из бактериофагов SP6, Т7 и ТЗ. Более подробную информацию по синтезу РНК-зондов можно получить из оригинальных публикаций, посвященных системе SP6.
Чтобы предупредить нежелательные последствия активности чужеродных рибонуклеаз, следует соблюдать определенные меры предосторожности при приготовлении растворов, при работе с пробирками, автоматическими пипетками и т.д. Растворы, выдерживающие нагревание, необходимо автоклавировать и хранить в стерильном виде. Бычий сывороточный альбумин, дитиотрейтол и другие растворы, не выдерживающие автоклавирования, следует готовить на стерильной воде и хранить в стерильных пробирках или бутылях. При работе с РНК желательно также стерилизовать пластиковую посуду и регулярно чистить стержни автоматических пипеток. 1. Матричная ДНК. Фрагмент ДНК-мишени нужно клонировать в плазмидном или фаговом векторе в участке, примыкающем к высокоспецифическим последовательностям промотора бактериофага. В качестве векторных систем обычно используются фаги SP6, Т7 и ТЗ. Имеются также векторы с одним либо двумя промоторами, обращенными к полилин-керным клонирующим сайтам, например pSP70-cepии, pGEM-серии, pBluescribe-серии. В идеальном случае последовательность-мишень встраивается в полилинкер между промоторами так, чтобы обратные РНК-транскрипты можно было получать с обеих ее цепей. После клонирования последовательности-мишени в векторной плазмиде с двумя промоторами обработайте 10тАУ20 мкг плазмидной ДНК рестриктазой, разрезающей по одному из концов мишени или внутри полилинкерной последовательности. В результате вы получите матрицу для обратной транскрипции. Если в качестве зонда предстоит использовать обе цепи матричной ДНК, обработайте такое же количество ДНК еще одной рестриктазой, разрезающей по противоположному концу последовательности-мишени. Экстрагируйте обработанную рестриктазами ДНК фенолом, осадите этанолом и перерастворите в ТЕ в концентрации 1000 мкг/мл.
2. РНК-полимеразы. Очищенные РНК-полимеразы из SP6, Т7 и ТЗ выпускаются многими фирмами. Поступающие в продажу препараты обычно имеют концентрацию 5тАУ20 ед./мл.
3. Меченые нуклеотиды. Приобретите один из четырех нуклеозидтрифосфатов, меченный фосфором в альфа-положении с удельной активностью 400 Ки/ммоль. Мы рекомендуем меченый GTP и приводим методику получения зонда на основе именно этого нуклеотида. Можно одновременно использовать и еще один меченый NTP, изменив соответственно количество взятых в реакцию немеченых нуклеозидтрифосфатов.
4. Немеченый GTP. 2 мм раствор ВлхолодногоВ» GTP хранить при тАУ 70В°С.
5. Смесь 3-NTP. Для приготовления Вл3-NTPВ» используют 50 или 100 мм исходные растворы остальных трех нуклеотидов в ТЕ-буфере. Концентрация каждого NTP в смеси 10 мм. Хранить при тАУ 70В°С.
6. Для транскрипции. Для приготовления 1 мл буфера необходимо:
400 мм трис-HCl, рН 7,5 400 мкл 1 М раствора 60 мм MgCl2 60 мкл 1 М раствора
20 мм спермидина 200 мкл 100 мм раствора 340 мкл Н20
7. Исходный раствор DTT. Приготовьте 1,0 М исходный раствор DTT в воде и храните его при 20оС.
8. Исходный раствор БСА. Приготовьте водный раствор без РНКазного БСА в концентрации 1 мг/мл и храните при тАУ20В°С.
9. Плацентный ингибитор РНКазы. Плацентный ингибитор РНКазы производится различными фирмами, в том числе и фирмой Promega Biotec Inc.
10. ДНКаза I. Приобретите сверхчистую, свободную от РНКазы ДНКазу I и приготовьте водный раствор в концентрации 1 мг/мл. Храните в аликвотах при тАУ 20В°С или тАУ70В°С. В своей работе мы пользовались ДНКазой фирмы Cooper Biochemical.
11. ДСН. Приготовьте 25%-ный исходный раствор ДСН в воде. Если есть возможность, используйте реактив ВлсверхчистойВ» квалификации. Не автоклавируйте. Храните при комнатной температуре.
12. тРНК-носитель. На некоторых этапах процесса требуется тРНК-носитель, свободный от примесей РНКазы и ДНКазы. Мы рекомендуем использовать дрожжевую тРНК, тщательно очищенную следующим образом:
а) суспендируйте тРНК в ТЕ из расчета 20 мг/мл, добавьте ДСН до 0,1% и протеиназу К до конечной концентрации 100 кмг/мл;
б) инкубируйте при 37В°тАУ50В°С в течение ночи;
в) экстрагируйте 3тАУ4 раза фенолом и диализуйте в течение ночи против нескольких литров ТЕ;
г) доведите концентрацию до 5 мг/мл и храните в аликвотах при тАУ70В°С.
4.1.2 Реактивы для отжига, расщепления и анализа гелей
Для отжига, реакций расщепления и анализа гелей в процессе РНКазного расщепления требуются следующие реактивы:
1. Тестируемая ДНК тАУ Тестировать можно геномную или клонированную ДНК, амплифицированную в ПЦР, просто клонированную и неамплифицированную геномную ДНК тАУ Получение ДНК в ПЦР описано в разд. 4. Клонированную и неамплифицированную геномную ДНК следует предварительно обработать рестриктазами для последующего количественного учета, а также для достижения максимально эффективной денатурации и реассоциации с зондом. Обработайте ДНК рестриктазами, выщепляющими тестируемый фрагмент, удалите ферменты фенольной экстракцией, сконцентрируйте ДНК, осадив ее этанолом. Ресуспендируйте в ТЕ-буфере в концентрации 100 мкг/мл.
2. Буфер для гибридизации. Для приготовления 10 мл необходимо: 80% формамида 8 мл деионизованного формамида 50 мм PIPES буфера, 1 мл 0,5 М PIPES, рН 6,4 рН
Не автоклавируйте Примечание: приобретайте формамид высокого качества и деионизируйте, осторожно перемешивая с ионообменной смолой Dowex AG50W или ее аналогами в течение 30 мин при комнатной температуре. Используйте примерно 2 г смолы на 100 мл формамида. Соберите формамид, профильтровав его через ватмановскую бумагу 1 ММ и храните в плотно закрытых пробирках при тАУ20В°С или тАУ70В°С.
Некоторые партии формамида содержат примеси, разрушающие РНК, особенно при высоких температурах. Это приводит к появлению на радиоавтографах высокого фона продуктов реакции РНКазного расщепления. Если остро встает проблема с фоном, рекомендуем перекристаллизовывать формамид перед использованием.
4.2 Методы
4.2.1 Синтез РНК-зондов
Описывается синтез РНК-зондов путем транскрипции in vitro клонированных ДНК, несущих фаговый промотор. Заметим, что удельная активность таких РНК-зондов ниже, чем полученных стандартным способом и традиционно используемых при тестировании геномных ДНК. Такой активности достаточно для работы с одной десятой от общего количества ДНК, получаемой в ПЦР, или с очень небольшим объемом клонированной ДНК тАУ Преимущество этих зондов состоит в том, что они сохраняют стабильность более 1 нед. Кроме того, длинные зонды с выступающими концами легче синтезировать так как при этом нуклеозидтрифосфаты не лимитируют реакцию транскрипции. Если исследуется геномная ДНК и не проводится ПЦР, то следует использовать зонды с высокой удельной активностью. Важно, чтобы концентрация меченого NTP в начале транскрипции была не менее 10 мкМ, поэтому для синтеза высокоактивного зонда требуется 40тАУ80 мкКи меченого NTP.
1. В стерильной микроцентрифужной пробирке смешайте в следующем порядке:
9 мкл воды,
1 мкл рестрицированной матричной ДНК,
1 мкл смеси 3-NTP,
1 мкл ВлхолодногоВ» GTP,
1 мкл GTP,
2 мкл DTT,
2 мкл БСА,
2тАУ5 единиц плацентного ингибитора РНКазы, 2 мкл 10Х буфера для транскрипции.
2. К смеси добавьте 3тАУ5 единиц РНК-полимеразы фагов SP6, Т7 или ТЗ.
3. Инкубируйте 1 ч при 40В°С, чтобы прошла транскрипция.
4. Добавьте 1 мкл ДНКазы I для разрушения матричной ДНК; инкубируйте 20 мин при 37? С.
5. Добавьте 10 мкг тРНК-носителя, 10 мкл воды и 20 мкл ацетата аммония и проэкстрагируйте один раз 2FC.
6. После экстракции водный слой осадите этанолом; ресуспендируйте осадок в 80 мл воды, 20 мл ацетата аммония и снова осадите этанолом.
7. Ресуспендируйте осадок РНК в 200 мкл ТЕ-буфера, содержащего 0,1% ДСН. Храните при тАУ20В°С.
4.2.2 Реакции отжига и расщепления
Первоначально реакцию гибридизации между меченым РНК-зондом и небольшим количеством комплементарных последовательностей из нескольких микрограмм суммарной геномной ДНК проводили с молярным избытком РНК тАУ Сейчас, когда за счет амплификации ДНК в ПЦР удается получать большие количества специфических фрагментов ДНК, нет необходимости использовать избыток зонда. В описываемых ниже экспериментах используется по существу избыток тестируемой ДНК тАУ Эта модификация позволяет увеличить отношение сигнала к фону, так как в этом случае уже не приходится удалять остатки длинного зонда с помощью РНКазной обработки.
При постановке эксперимента полезно иметь следующие контроли:
контроль 1: неспецифическая гибридизация зонда с тРНК-носителем;
контроль 2: положительный контроль на дикий тип тАУ реассоциация зонда с клонированным фрагментом ДНК дикого типа;
контроль 3: зонд реассоциируют с известным мутантным фрагментом геномной ДНК, клонированным или полученным в ПЦР. Это дает возможность оценить эффективность расщепления неспарен, ных участков.
1. В стерильной микроцентрифужной пробирке смешайте следующие компоненты:
1 мкл амплифицированной в ПЦР геномной ДНК в концентрации примерно 100 мкг/мл или 1 мкл рестрицированной клонированной ДНК,
1 мкл меченого РНК-зонда, 30 мкл буфера для гибридизации.
2. Прогрейте смесь 10 мин при 95тАУ100В°С на кипящей водяной бане.
3. Воспользуйтесь микроцентрифугой, чтобы собрать конденсат на дне пробирки.
4. Инкубируйте смесь 60 мин при 40тАУ45В°С, чтобы произошла реассоциация между зондом и комплементарными ему последовательностями в тестируемом фрагменте геномной ДНК.
Примечание: по оригинальной методике гибридизацию проводят в течение 10тАУ12 ч. Считается, что за это время реакция реассоциации между фрагментами геномной ДНК и РНК-зондом достигает насыщения. В клонированных и амплифицированных в ПЦР фрагментах геномной ДНК доля последовательностей-мишеней намного больше, поэтому время гибридизации можно значительно уменьшить. Сокращение сроков инкубации облегчает проведение эксперимента и уменьшает деградацию зонда. 5. Добавьте 350 мкл смеси РНКазы с РНКазным буфером. Как следует перемешайте встряхиванием. Эта смесь представляет собой буфер для РНКазной реакции с разведенной в нем до концентрации 40 мкг/мл прокипяченной РНКазой А и тРНК-носителем в концентрации 20 мкг/мл. Раствор можно приготовить за несколько часов до использования и хранить во льду. На 5 мл смеси надо 5 мл буфера для РНКазной реакции, 100 мкл исходного раствора прокипяченной РНКазы А, 20 мкл тРНК. Примечание: в большинстве наших опытов по РНКазному расщеплению концентрация РНКазы А была 40 мкг/мл. Однако, используя фермент из другой партии, мы обнаружили, что эта концентрация слишком велика тАУ фермент в таком количестве разрушал гибриды РНК тАУ ДНК тАУ Положительных результатов удалось достичь, снизив его концентрацию до 4 мкг/мл. Поэтому мы рекомендуем каждую новую партию РНКазы перед использованием калибровать в пределе концентраций от 2 до 40 мкг/мл. Для калибровки желательно использовать как мутантный контрольный образец, так и контрольный образец дикого типа. В оптимальной концентрации РНКаза А эффективно расщепляет неспаренные участки в мутантном образце и дает фоновый сигнал в контроле дикого типа.
6. Инкубируйте 60 мин при 25В°С.
Примечание: по оригинальной методике инкубацию проводили в течение 30 мин. За это время РНКаза А расщепляет многие
Вместе с этим смотрят:
G-белки и их функция
Австралопитеки - обезьянолюди или человекообезьяны?
Адаптация микроорганизмов в экстремальных условиях космоса
Адвентивна флора Чернiгiвськоi областi: iсторiя формування та сучасний стан
Адсорбция ионных и неионных поверхностно-активных веществ (ПАВ)