Характеристика генных заболеваний
Проблема здоровья людей и генетика тесно взаимосвязаны. Ученые-генетики пытаются ответить на вопрос, почему одни люди подвержены различным заболеваниям, в то время как другие в этих или даже худших условиях остаются здоровы. В основном это связано с наследственностью каждого человека, т.е. свойствами его генов, заключенных в хромосомах.
В последние годы отмечаются быстрые темпы развития генетики человека и медицинской генетики. Это объясняется многими причинами и прежде всего резким увеличением доли наследственной патологии в структуре заболеваемости и смертности населения. Статистика показывает, что из 1000 новорожденных у 35-40 выявляются различные типы наследственных болезней, а в смертности детей в возрасте до 5 лет хромосомные болезни составляют 2-3%, генные тАФ 8-10%, мультифакторные тАФ 35-40%. Ежегодно в нашей стране рождается 180 тыс. детей с наследственными заболеваниями. Более половины из них имеют врожденные пороки, около 35тыс. тАФ хромосомные болезни и свыше 35 тыс. тАФ генные болезни. Следует отметить, что число наследственных болезней у человека с каждым годом растет, отмечаются новые формы наследственной патологии. В 1956 г. было известно 700 форм наследственных заболеваний, а к 1986 году число их увеличилось до 2000. В 1992 количество известных наследственных болезней и признаков возросло до 5710.
Все наследственные болезни делятся на три группы:
1. Генные (моногенные тАУ в основе патологии одна пара аллельных генов).
2. Хромосомные.
3. Болезни с наследственным предрасположением (мультифакториальные)
ГЕННЫЕ БОЛЕЗНИ
Генные болезни тАУ это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена.
Общая частота генных болезней в популяции составляет 1-2 %. Условно частоту генных болезней считают высокой, если она встречается с частотой 1 случай на 10.000 новорожденных, средней тАУ 1 на 10.000-40.000 и далее тАУ низкой.
Моногенные формы генных заболеваний наследуются в соответствии с законами Г. Менделя. По типу наследования они делятся на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с X- или Y-хромосомами.
Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов тАУ белков. Любая мутация гена ведет к изменению структуры или количества белка.
Начало любой генной болезни связано с первичным эффектом мутантного аллеля. Основная схема генных болезней включает ряд звеньев: мутантный аллель à измененный первичный продукт à цепь последующих биохимических процессов клетки à органы à организм.
В результате мутации гена на молекулярном уровне возможны следующие варианты:
1) синтез аномального белка;
2) выработка избыточного количества генного продукта;
3) отсутствие выработки первичного продукта;
4) выработка уменьшенного количества нормального первичного продукта.
Не заканчиваясь на молекулярном уровне в первичных звеньях, патогенез генных болезней продолжается на клеточном уровне. При различных болезнях точкой приложения действия мутантного гена могут быть как отдельные структуры клетки тАФ лизосомы, мембраны, митохондрии, пероксисомы, так и органы человека. Клинические проявления генных болезней, тяжесть и скорость их развития зависят от особенностей генотипа организма (гены-модификаторы, доза генов, время действия мутантного гена, гомо- и гетерозиготность и др.), возраста больного, условий внешней среды (питание, охлаждение, стрессы, переутомление) и других факторов.
Особенностью генных (как и вообще всех наследственных) болезней является их гетерогенность. Это означает, что одно и то же фенотипическое проявление болезни может быть обусловлено мутациями в разных генах или разными мутациями внутри одного гена. Впервые гетерогенность наследственных болезней была выявлена С.Н. Давиденковым в 1934 г.
К генным болезням у человека относятся многочисленные болезни обмена веществ. Они могут быть связаны с нарушением обмена углеводов, липидов, стероидов, пуринов и пиримидинов, билирубина, металлов и др. Пока еще нет единой классификации наследственных болезней обмена веществ. Научной группой ВОЗ предложена следующая классификация:
1) болезни аминокислотного обмена (фенилкетонурия, алкаптонурия и др.);
2) наследственные нарушения обмена углеводов (галагоземия, гликогеновая болезнь и др.);
3) болезни, связанные с нарушением липидного обмена (болезнь Ниманна-Пика, болезнь Гоше и др.);
4) наследственные нарушения обмена стероидов;
5) наследственные болезни пуринового и пиримидинового обмена (подагра, синдром Леша-Найяна и др.);
6) болезни нарушения обмена соединительной ткани (болезнь Марфана, мукополисахариды и др.);
7) наследственные нарушения гема- и порфирина (гемоглобинопатии и др.);
8) болезни, связанные с нарушением обмена в эритроцитах (гемолитические анемии и др.);
9) наследственные нарушения обмена билирубина;
10) наследственные болезни обмена металлов (болезнь Коновалова-Вильсона и др.);
11) наследственные синдромы нарушения всасывания в пищеварительном тракте (муковисцидоз, непереносимость лактозы и др.).
Рассмотрим наиболее часто встречающиеся и генетически наиболее изученные в настоящее время генные болезни.
Наследственные болезни аминокислотного обмена
Это тАУ самая многочисленная группа наследственных болезней обмена веществ. Почти все они наследуются по аутосомно-рецессивному типу. Причина заболеваний тАУ недостаточность того или иного фермента, ответственного за синтез аминокислот. Болезни сопровождаются рвотой и обезвоживанием организма, летаргическим состоянием или возбуждением и судорогами. В позднем возрасте проявляется угасание умственного и физического развития.
К наследственным болезням с нарушенным аминокислотным обменом относится фенилкетонурия, альбинизм и др.
Фенилкетонурия (ФКУ) впервые была описана А. Фелингом в 1934 г. У больных нарушено превращение аминокислоты фенилаланина в тирозин из-за резкого снижения активности фермента фенилаланингидроксилазы. В результате содержание фенилаланина в крови и моче больных значительно возрастает. Далее фенилаланин превращается в фенилпировиноградную кислоту, которая является нейротропньм ядом и нарушает формирование миелиновой оболочки вокруг аксонов центральной нервной системы.
Фенилкетонурия встречается в среднем в мировом масштабе с частотой 1 на 1000 новорожденных. Однако по этому показателю имеются значительные различия между популяциями: 1:2600 в Турции, 1:4500 в Ирландии, 1: 30000 в Швеции, 1:119000 в Японии. Частота гетерозиготного носительства в большинстве европейских популяций составляет 1:100.
Локус (фенилгидроксилазы) расположен в длинном плече 12-й хромосомы. В настоящее время для большинства семей возможна молекулярно-ге-нетическая диагностика и выявление гетерозиготного носительства. Болезнь наследуется по аутосомно-доминантному типу. Известно несколько форм фенилкетонурии, которые различаются по тяжести протекания болезни. Это связано с наличием 4-х аллелей гена и их комбинациями.
Ребенок с фенилкетонурией рождается здоровым, но в первые же недели в связи с поступлением фенилаланина в организм с молоком матери развивается повышенная возбудимость, судорожный синдром, склонность к дерматитам, моча и пот больных имеют характерный ВлмышиныйВ» запах, но главными симптомами ФКУ являются судорожные припадки и олигофрения.
Большинство больных - блондины со светлой кожей и голубыми глазами, что определяется недостаточным синтезом пигмента меланина. Диагноз заболевания устанавливается на основании клинических данных и результатов биохимического анализа мочи (на фенилпировиноградную кислоту) и крови (на фенилаланин). С этой целью несколько капель крови на фильтровальной бумаге подвергают хроматографии и определяют содержание фенилаланина. Иногда используют пробу Феллинга тАУ в 2,5 мл свежей мочи ребенка добавляют 10 капель 5% раствора треххлористого железа и уксусной кислоты. Появление сине-зеленого окрашивания указывает на наличие заболевания.
Метод лечения фенилкетонурии в настоящее время хорошо разработан. Он состоит в назначении больному диеты (овощи, фрукты, варенье, мед) и специально обработанных гидролизатов белков с низким содержанием фенилаланина (лофелак, кетонил, минафен и др). В настоящее время разработаны методы дородовой диагностики. Ранняя диагностика и профилактическое лечение предупреждают развитие болезни.
Альбинизм (глазо-кожный) описан в 1959 г. Болезнь обусловлена отсутствием фермента тирозиназы. Для нее характерна обесцвеченность кожи, волос, глаз, независимо от расы и возраста. Кожа больных розово-красная, совершенно не загорает. Имеет предрасположенность к злокачественным новообразованиям. Волосы белые или желтоватые. Радужка серо-голубого цвета, но может быть и розоватая из-за отражения света от глазного дна. Больным свойственна сильная светобоязнь, их зрение снижено и не улучшается с возрастом.
Альбинизм встречается с частотой 1 на 39.000, наследуется по аутосомно-рецессивному типу (см. приложение рис. 1). Ген локализован на длинном плече 11-й хромосомы.
Наследственные заболевания, связанные с нарушением обмена углеводов
Известно, что углеводы входят в состав ряда биологически-активных веществ тАФ гормонов, ферментов, мукополисахаридов, выполняющих энергетическую и структурную функции. В результате нарушения углеводного обмена развивается гликогеновая болезнь, галактоземия и др.
Гликогеновая болезнь связана с нарушением синтеза и разложения гликогена тАФ животного крахмала. Гликоген образуется из глюкозы при голодании; в норме он снова превращается в глюкозу и усваивается организмом. При нарушении этих процессов у человека развиваются тяжелые заболевания тАФ различные типы гликогенозов. К ним относятся болезнь Гирке, болезнь Помпе и др.
Гликогеноз (I тип тАФ болезнь Гирке). У больных в печени, почках и слизистой кишечника накапливается большое количество гликогена. Превращение его в глюкозу не происходит, т.к. отсутствует фермент глюкоо-6-фосфатаза, регулирующий уровень глюкозы в крови. В результате у больного развивается гипогликемия, в печени, почках и слизистой кишечника накапливается гликоген. Болезнь Гирке наследуется по аутосомно-рецессивному типу.
Сразу после рождения главными симптомами болезни являются гликогемические судороги и гепатомегалия (увеличение печени). С 1-го года жизни отмечается задержка роста. Характерен вид больного: большая голова, Влкукольное лицоВ», короткая шея, выступающий живот (см. приложение рис. 2). Кроме того, отмечаются носовые кровотечения, задержка физического и полового развития, мышечная гипотония. Интеллект при этом нормальный. В крови повышается уровень мочевой кислоты, так что с возрастом может развиться подагра.
В качестве лечения используется диетотерапия: частый прием пищи, повышенное содержание углеводов и ограничение жиров в диете.
Глнкогеноз (II тип тАФ болезнь Помпе) протекает в более тяжелой форме. Гликоген накапливается как в печени, так и в скелетных мышцах, миокарде, легких, селезенке, надпочечниках, стенках сосудов, в нейронах.
У новорожденных спустя 1-2 месяца появляется мышечная слабость, дефицит 1,4-глюкозидазы в печени и в мышцах. В этот же период возникают кардиомегалия (увеличение сердца) и макроглоссия (патологическое увеличение языка). Нередко у больных развивается тяжелая форма пневмонии из-за накопления секрета в дыхательных путях. Дети погибают на первом году жизни.
Болезнь наследуется по аутосомно-рецессивному типу. Ген локализован на длинном плече 17-й хромосомы. Диагностика заболевания возможна еще до рождения ребенка. С этой целью определяют активность фермента 1,4-глюкозидазы в амниотической жидкости и ее клетках.
Известны и другие типы гликогенозов (III-VII), которые в общем повторяют клиническую картину первого типа. Для их диагностики необходимы специальные биохимические исследования.
Галактоземия. При этом заболевании происходит накопление в крови больного галактозы, что приводит к поражению многих органов: печени, нервной системы, глаз и др. Симптомы болезни появляются у новорожденных после приема молока, поскольку галактоза тАУ составная часть молочного сахара лактозы. При гидролизе лактозы образуется глюкоза и галактоза. Последняя необходима для миелинизации нервных волокон. При избытке галактозы в организме она в норме превращается в глюкозу с помощью фермента галактоза-1-фосфат-уридилтрансферазы. При понижении активности этого фермента происходит накопление галактоза-1-фосфата, токсичного для печени, мозга, хрусталика глаза.
Болезнь проявляется с первых дней жизни расстройствами пищеварения, интоксикацией (понос, рвота, обезвоживание). У больных увеличивается печень, развивается печеночная недостаточность и желтуха. Обнаруживается катаракта (помутнение хрусталика глаза), умственная отсталость. У погибших в первый год жизни детей при вскрытии обнаруживают цирроз печени.
Наиболее точные методы диагностики галактоземии тАУ определение активности фермента галактоза-1-фосфат-уридилтрансферазы в эритроцитах, а также галактозы в крови и моче, где уровни ее увеличены. При исключении из пищи молока (источника галактозы) и раннем назначении диеты больные дети могут нормально развиваться.
Тип наследования галактоземии тАФ аутосомно-рецессивный. Ген локализован па коротком плече 9-й хромосомы. Болезнь встречается с частотой 1 на 16.000 новорожденных.
Наследственные заболевания, связанные с нарушением липидного обмена
В число липидов входят холестерол (холестерин), триглицериды, эфиры холестерола, фосфолипиды, сфинголипиды и др.
Наследственные болезни обмена липидов (липидозы) подразделяются на два основных типа: 1) внутриклеточные, при которых происходит накопление липидов в клетках различных тканей и 2) болезни с нарушением метаболизма липопротеидов, содержащихся в крови.
К числу наиболее изученных наследственных заболеваний липидного обмена первого типа относятся болезнь Гоше, болезнь Ниманна-Пика и амавротическая идиотия (болезнь Тея-Сакса).
Болезнь Гоше характеризуется накоплением цереброзидов в клетках нервной и ретикуло-эндотелиальной системы, обусловленным дефицитом фермента глюкоцереброзидазы. Это приводит к накоплению в клетках ретикуло-эндотелиальной системы глюкоцереброзида. В клетках мозга, печени, лимфатических узлах обнаруживаются крупные клетки Гоше. Накопление цереброзида в клетках нервной системы приводит к их разрушению.
Выделяют детскую и ювенильную формы болезни. Детская проявляется в первые месяцы жизни задержкой умственного и физического развития, увеличением живота, печени и селезенки, затруднением глотания, спазмом гортани. Возможна дыхательная недостаточность, инфильтрация (уплотнение легких клетками Гоше) и судороги. Смерть наступает на первом году жизни (см. приложение рис. 3).
Наиболее часто встречается ювенильная форма болезни Гоше. Она поражает детей различного возраста и носит хронический характер. Заболевание проявляется, как правило, на первом году жизни. Возникают пигментация кожи (коричневые пятна), остеопороз (снижение плотности кости), переломы, деформация костей. В тканях мозга, печени, селезенки, костного мозга содержится большое количество глюкоцереброзидов. В лейкоцитах, клетках печени и селезенки снижена активность глюкозидазы. Тип наследования аутосомно-рецессивный. Ген локализован на длинном плече 1-й хромосомы.
Болезнь Ниманн - Пика обусловлена снижением активности фермента сфингомиелиназы. В результате происходит накопление сфингомиелина в клетках печени, селезенке, мозге, ретикуло тАУ эндотелиальной системе. Вследствие дегенерации нервных клеток нарушается деятельность нервной системы.
Выделяют несколько форм заболевания, различающихся клинически (время начала, течение и тяжесть неврологических проявлений). Однако имеются и общие для всех форм симптомы.
Болезнь чаще проявляется в раннем возрасте. У ребенка увеличиваются лимфатические узлы, размеры живота, печени и селезенки; отмечаются рвота, отказ от пищи, мышечная слабость, снижение слуха и зрение (см. приложение рис. 4). У 20-30% детей на сетчатке глаза обнаруживается пятно вишневого цвета (симптом Влвишневой косточкиВ»). Поражение нервной системы ведет к отставанию нервно-психического развития, глухоте, слепоте. Резко снижается устойчивость к инфекционным заболеваниям. Дети погибают в раннем возрасте.
Наследование болезни тАУ аутосомно-рецессивное. Ген сфингомиелиназы картирован на хромосоме 11.
Диагностика болезни Ниманна-Пика основана на выявлении в плазме крови и спинномозговой жидкости повышенного содержания сфингомиелина. В периферической крови выявляются большие зернистые пенистые клетки Пика. Лечение симптоматическое.
Амавротическая идиотия (болезнь Тея-Сакса) также относится к заболеваниям, связанным с нарушением липидного обмена. Для нее характерно отложение в клетках мозга, печени, селезенки и других органах липида ганглиозида. Причина тАУ снижение активности фермента гексозаминидазы А в организме. В результате происходит разрушение аксонов нервных клеток.
Болезнь проявляется в первые месяцы жизни. Ребенок становится вялым, малоподвижным, безразличным к окружающим. Задержка психического развития приводит к снижению интеллекта до степени идиотии. Отмечается мышечная гипотония, судороги, характерный симптом "вишневой косточки" на сетчатке глаза. К концу первого года жизни наступает слепота. Причина тАФ атрофия зрительных нервов. Позднее развивается полная обездвиженность. Смерть наступает в 3-4 года.
Тип наследования болезни тАФ аутосомно-рецессивный. Ген локализован на длинном плече 15-й хромосомы.
Наследственные болезни соединительной ткани
Роль соединительной ткани в организме связана с опорной, трофической и защитной функциями. Важнейшими компонентами, соединительной ткани являются:
а)ВаВаВаВаВа клеточные компоненты;
б)ВаВаВаВаВа коллагеновые, эластические и ретикулярные волокна;
в)ВаВаВаВаВа аморфное основное вещество.
Сложная структура соединительной ткани задана генетически. Патология в
ее системе является причиной различных наследственных заболеваний и обусловлена в той или иной степени нарушениями строения структурных белков тАФ коллагенов.
Большинство болезней соединительной ткани связано с дефектами опорно-двигательного аппарата и кожи. К числу их относятся синдром Марфана, синдром Элерса-Данлоса, а также мукополисахаридозы.
Синдром Марфана ("паучьи пальцы") относится к числу наследственных болезней обмена веществ и характеризуется системным поражением соединительной ткани. Наследуется по аутосомно-доминантному типу с высокой пенетрантностью и различной степенью экспрессивности. С этим связан значительный клинический и возрастной полиморфизм. Впервые синдром был описан В. Марфаном в 1886 г. Причина болезни тАФ мутация в гене, ответственном за синтез белка соединительнотканных волокон фибриллина. Блокирование его синтеза приводит к повышенной растяжимости соединительной ткани.
Больных с синдромом Марфана отличают высокий рост, длинные паукообразные пальцы, деформация грудной клетки (воронкообразная, килевидная, уплощенная), плоскостопие. Нередко имеют место бедренные и паховые грыжи, гипоплазия (недоразвитие) мышц, мышечная гипотония, ухудшение зрения, изменение формы и размера хрусталика, значительная миопия вплоть до отслойки сетчатки, гетерохромия (разное окрашивание участков радужки); подвывих хрусталика, катаракта, косоглазие.
Помимо перечисленного, при синдроме Марфана характерны врожденные пороки сердца, расширение аорты с развитием аневризмы. Нередко отмечаются расстройства органов дыхания, поражения желудочно-кишечного тракта и мочевыводящей системы.
Лечение в основном симптоматическое. Положительное действие оказывают массаж, лечебная физкультура, а в ряде случаев оперативное вмешательство. Большое значение имеет ранняя диагностика заболевания. Частота синдрома Марфана в популяции равна 1:10.0(1:15.000).
Следует отметить, что синдромом Марфана страдали президент США Авраам Линкольн, великий итальянский скрипач и композитор Николо Паганини.
Мукополисахаридозы представлены целой группой наследственных заболеваний соединительной ткани. Для них характерно нарушение в организме метаболизма кислых гликозаминогликанов, что связано с недостаточностью лизосомальных ферментов. В результате патологические продукты обмена откладываются в соединительной ткани, в печени, селезенке, роговице и в клетках центральной нервной системы.
Первые сведения о мукополисахаридозах появились в 1900 г., а затем в 1947-1919 гг. Еще раньше это заболевание было известно под названием "гаргоилизм", поскольку внешне пробанды напоминали фигуры, украшавшие собор Парижской Богоматери.
При мукополисахаридозах поражаются опорно-двигательный аппарат, внутренние органы, глаза, нервная система.
Клиническими признаками болезни служат замедление роста, короткая шея и туловище, деформация костей, снижение интеллекта, грубые черты лица с крупными губами и языком, пупочные и паховые грыжи, пороки сердца, нарушение психического развития с отставанием от нормы.
Тип наследования болезни тАФ аутосомно-рецессивный. Ген картирован на коротком плече 4-к хромосомы.
Всего выделяют 8 основных типов мукополисахаридозов в зависимости от снижения активности разных ферментов и особенностей клинических признаков. Для определения типа заболевания исследуются биохимические показатели кислых гликозаминоглюканов в крови и моче больных.
Лечение: диетотерапия, физиопроцедуры (электрофорез, магнитотера-пия, массаж, лечебная физкультура и др.), гормональные и сердечно-сосудистые средства.
Наследственные нарушения обмена в эритроцитах
К этой группе относятся болезни, связанные чаще всего с укорочением срока жизни эритроцитов, а также со снижением его уровня в крови. Гемоглобин тАФ основной белок эритроцитов. В настоящее время хорошо изучена аминокислотная последовательность и структура его молекулы. Изучение гемоглобина началось с открытия наследственного заболевания тАУсерповидноклеточной анемии. Было показано, что молекулярная структура серповидноклеточного гемоглобина отличается от нормального.
По мере совершенствования методов электрофореза были выявлены более 400 вариантов гемоглобина и расшифрована их полная аминокислотная последовательность. Установлено, что молекула человеческого гемоглобина состоит из 4-х полипептидных цепей (α, β, γ, δ). Большинство разновидностей гемоглобина человека имеет идентичные α-цепи и различаются по остальным цепям. К каждой цепи глобина в специфическом участке присоединяется молекула небелковой природы тАУ гемогруппы, или гемм. Четыре глобиновых цепи вместе с геммами образуют функциональную молекулу гемоглобина, которая переносит кислород из легких в ткани.
Молекула глобина состоит из 141 тАУ 146 аминокислотных остатков, расположенных в строго определенном порядке. Последовательность аминокислот в белке называется его первичной структурой. Пространственное взаиморасположение соседних мономеров в полипептидной цепи именуется вторичной структурой, а трехмерная конфигурация белковых субъединиц тАУ структурой третичной. Четвертичная же структура гемоглобина реализуется во взаимной организации четырех субъединиц в составе функционирующей молекулы.
У детей и взрослых преобладающим типом гемоглобина является НвА, состоящий из двух α- и двух β-цепей (a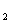 β), α-цепи у всех гемоглобинов идентичны. α- и β-цепи различаются по многим аминокислотным остаткам. У всех взрослых есть 2-3 % гемоглобина НвА2. Характерная для него δ -цепь отличается от β -цепи по 10 аминокислотным остаткам. После рождения у всех детей обнаруживается также небольшое количество {меньше 1%) фетального гемоглобина НbF. γ тАУ цепь значительно отличается от β -цепей. Синтез γ -цепей у эмбриона происходит в основном в печени и селезенке, но могут они синтезироваться и в клетках костного мозга. А β -цепи, наоборот, синтезируются главным образом в клетках костного мозга.
β), α-цепи у всех гемоглобинов идентичны. α- и β-цепи различаются по многим аминокислотным остаткам. У всех взрослых есть 2-3 % гемоглобина НвА2. Характерная для него δ -цепь отличается от β -цепи по 10 аминокислотным остаткам. После рождения у всех детей обнаруживается также небольшое количество {меньше 1%) фетального гемоглобина НbF. γ тАУ цепь значительно отличается от β -цепей. Синтез γ -цепей у эмбриона происходит в основном в печени и селезенке, но могут они синтезироваться и в клетках костного мозга. А β -цепи, наоборот, синтезируются главным образом в клетках костного мозга.
Все нормальные гемоглобины человека имеют идентичную трехмерную структуру, оптимальную для переноса кислорода.
Аминокислотная последовательность каждой глобиновой цепи кодируется своим геном, равно как и синтез небелковой гемогруппы.
Гены глобинов у человека образуют мультигенное семейство и расположены на хромосомах двух кластеров (Кластеры тАФ группа генов, расположенных в определенных локусах, объединенных общими функциями.) тАФ α -кластер занимает 25000 пар нуклеотидов в коротком плече 16-й хромосомы. Семейство γ - β - δ -генов расположено в коротком плече 11-й хромосомы (60 000 п. н.).
Все глобиновые гены имеют сходную функциональную организацию. Они содержат 3 кодирующие последовательности (зкзоны) и 2 интрона, которые транскрибируются вместе с экзонами и вырезаются в ходе процессннга. Варианты гемоглобинов возникают в результате мутаций в конкретном глобиновом гене. Эти варианты (чаще всего) отличаются одной аминокислотой в глобиновой цепи. Описано более 350 таких единичных замен. Замены аминокислот влияют на сродство молекулы гемоглобина кислорода. Нарушение же его функций ведет к возникновению заболеваний. К другим типам мутаций, изменяющим гемоглобин, относятся: делеции, дупликации, мутации сдвига рамки считывания, мутации, нарушающие процессинг РНК и др.
В результате мутаций в эритроцитах и гемоглобине возникают наследственные болезни человека тАУ гемолитические анемии и гемоглобинопатии.
Гемолитические анемии включают заболевания, обусловленные снижением уровня гемоглобина и укорочением срока жизни эритроцитов. Кроме того, причиной болезни могут быть:
1) Нарушение мембраны эритроцитов.
2) Нарушение активности ферментов эритроцитов (ферментов, гликолиза пентозофосфатного цикла и др.).
3) Нарушение структуры или синтеза гемоглобина.
Рассмотрим кратко наиболее распространенную форму наследственной гемолитической анемии у человека.
Наследственный микросфероцитоз тАУ гемолитическая анемия Минковского-Шоффара. Болезнь описана в 1900 г. Примерно в половине случаев она проявляется у новорожденных. Диагноз ставится в возрасте 3-10 лет. Заболевание обусловлено генетическими аномалиями эритроцитов и связано с врожденной недостаточностью липидов их оболочки. В результате повышения проницаемости мембраны в клетку проникают ионы натрия и теряется АТФ. Эритроциты принимают сферическую форму. Измененные эритроциты разрушаются в селезенке с образованием токсического белка тАУ билирубина.
При данном заболевании отмечают желтуху, анемию, спленомегалию (разрыв селезенки), изменения скелета. Болезнь может протекать в двух формах тАФ хронической и острой, при которой усиливается гемолиз, обусловливающий анемию.
У детей в первые месяцы жизни часто возникает "ядерная желтуха". Причина тАФ поражение ядер головного мозга за счет высокого содержания билирубина. В более старшем возрасте высокий уровень билирубина приводит к образованию камней и развитию желчнокаменной болезни.
Для больных характерно увеличение селезенки и печени, деформация скелета; башенный череп, готическое небо, нарушенное расположение зубов.
Для диагностики заболевания исследуют кровь. Характерными признаками являются обнаружение микросфероцитов, снижение осмотической стойкости эритроцитов и др.
Тип наследования тАФ аутосомно-доминантный с неполной пенетрантностью. Ген картирован на коротком плече 8-й хромосомы.
Наследственные аномалии циркулирующих белков. Гемоглобинопатии.
Гемоглобинопатии тАФ это болезни, связанные с наследственным нарушением синтеза гемоглобина. Выделяют количественные (структурные) и качественные их формы. Первые характеризуются изменением первичной структуры белков гемоглобина, что может приводить к нарушению его стабильности и функции. При качественных формах структура гемоглобина остается нормальной, снижена лишь скорость синтеза глобиновых цепей.
Таласеемии. Данная патология обусловлена снижением скорости синтеза полипептидных цепей нормального гемоглобина А. Впервые бодазнь описана в 1925 г. Ее название происходит от греческого "Таласса" тАФ Средиземное море. Полагают, что со средиземноморским регионом связано происхождение большинства носителей гена талассемии.
Талассемия встречается в гомо- и гетерозиготных формах. По клинической картине принято различать большую, промежуточную, малую и минимальную формы. Остановимся на одной из них.
Гомозиготная (большая) талассемня, она же тАФ анемия Кули вызывается резким снижением образования гемоглобина НbA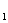 и увеличением количества гемоглобина F.
и увеличением количества гемоглобина F.
Клинически болезнь проявляется к концу первого года жизни ребенка. Для нее характерны монгодлоидность лица, башенный тип черепа, отставание физического развития. При данной патологии в крови больного обнаруживаются мишеневидные эритроциты с низким содержанием Нв, укороченной продолжительностью жизни и повышенной осмотической стойкостью. У больных отмечается увеличение селезенки и, реже, печени.
По тяжести заболевания выделяют несколько форм талассемии. Тяжелая талассемия заканчивается быстрой гибелью в первые месяцы жизни ребенка. При хронической тАФ больные дети доживают до 5-8 лет, а при легких формах, больные доживают до взрослого возраста,
Серповидноклеточная анемия тАФ наиболее часто встречаемое наследственное заболевание, вызванное изменением структуры молекулы гемоглобина. Люди, страдающие серповидно-клеточной анемией, в большинстве случаев погибают, не достигнув зрелого возраста. В условиях низкого парциального давления кислорода их эритроциты приобретают форму серпа. У родителей больного эритроциты имеют несколько измененную форму, но они не страдают анемией.
Это заболевание впервые обнаружил в 1910 г. Дж. Херрик у студента-негра, страдавшего тяжелой формой анемии. В крови больного он выявил эритроциты необычной серповидной формы. Оказалось, что это заболевание не редкое явление у американских негров (см. приложение рис. 8).
В 1946 г. Нобелевский лауреат Л. Полинг с коллегами провели биохимический и генетический анализ гемоглобина больных и здоровых людей и показали, что гемоглобины нормальных и серповидноклеточных эритроцитов различаются по подвижности в электрическом поле и растворимости. Оказалось, что гемоглобин у людей с признаками серповидноклеточности представляет смесь равных количеств и нормального и мутантного гемоглобина. Стало ясно, что мутация, вызывающая серповидноклеточную анемию, связана с изменением химической структуры гемоглобина.
Дальнейшие исследования показали, что в случае серповидноклеточной анемии происходит замена глютаминовой кислоты на валин в шестой паре нуклеотидов гена, кодирующего β-цепь гемоглобина человека. У гетеро-зигот измененный гемоглобин составляет 20-45 %, у гомозигот - 60-99 % общего гемоглобина.
При данной патологии отмечают бледность кожи и слизистых оболочек, желтушность. У 60% детей увеличена печень. Отмечается также шумы в области сердца и др. Болезнь протекает в виде чередования кризов и ремиссий.
Специальных методов лечения нет. Важное значение имеет предохранение больного от воздействия факторов, провоцирующих развитие болезни (гипоксия, обезвоживание, холод и др.).
ХРОМОСОМНЫЕ БОЛЕЗНИ ЧЕЛОВЕКА
К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом.
Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3-5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мертворождений. По данным 1987 г. из 1000 новорожденных 7 имели различные хромосомные аномалии.
Все хромосомные болезни принято делить на две группы.
Связанные с аномалиями числа хромосом.
В эту группу входят три подгруппы:
1.Болезни, обусловленные нарушением числа аутосом.
2.Болезни, связанные с увеличением или уменьшением числа половых Х- и У-хромосом.
3.Болезни, причиной которых является полиплоидия тАФ кратное увеличение гаплоидного набора хромосом.
Связанные со структурными нарушениями (аберрациями) хромосом.
Их причинами бывают:
1. Транслокации тАУ обменные перестройки между негомологичными хромосомами.
2. Делеции тАУ потери участка хромосомы.
3. Инверсии тАУ повороты участка хромосомы на 180 градусов.
4. Дупликации тАУ удвоения участка хромосомы.
5. Изохромосомия тАУ хромосомы с повторяющимися генетическим материалом в обоих плечах.
6. Возникновение кольцевых хромосом (соединение двух концевых делеций в обоих плечах хромосомы).
В настоящее время у человека известно более 700 таких заболеваний, вызванных структурными нарушениями хромосом. Имеющиеся данные показывают, что около 25% приходится на аутосомные трисомии, 46% тАФ на патологию половых хромосом. Структурные перестройки составляют 10,4%. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции.
Рассмотрим наиболее часто встречаемые хромосомные болезни.
Синдром Дауна (трисомия по 21-й хромосоме). Данное заболевание относится к числу наиболее распространенных патологий человека. Встречается у новорожденных с частотой 1:700-800. Частота рождения детей с болезнью Дауна зависит от возраста матери и в меньшей степени от возраста отца. Риск рождения больного ребенка матерью в 40-46 лет в 16 раз выше, чем в 20-24 года. Заметных различий по частоте заболевания болезнью Дауна в разных географических и расовых популяциях (см. приложение табл. 1 и 2) не отмечается. Обычно трисомия по 21-й паре хромосом составляет около 95% от общего числа больных с синдромом Дауна. Примерно 4% больных страдает транслокационной формой и около 2% тАФ мозичными. При транслокационном варианте в кариотипе наблюдается 46 хромосом, а лишняя 21-я хромосома транс-лоцирована чаще всего на хромосому из групп D или G.
Лежащее в основе синдрома Дауна нерасхождение 21-й пары хромосом происходит либо в яйцеклетке во время мейоза, либо на ранних стадиях дробления зиготы.
Соотношение больных мальчиков и девочек среди новорожденных составляет 1:1. Признаки синдрома заметны уже при рождении и позднее проявляются более четко. Больные обычно невысокого роста, отличаются слабоумием. У них характерная внешность: монголоидный разрез глаз, круглое уплощенное лицо, короткий нос, плоская переносица, эпикант (полулунная вертикальная складка у внутреннего угла глаза), маленькие деформированные уши, полуоткрытый рот га слегка высунутым языком и выступающей нижней челюстью (см. приложение рис. 9), (см. приложение табл. 3).
За счет сильной мышечной гипотонии объем движений в суставах увеличен. Дети с синдромом Дауна в первый год жизни заметно отстают в психомоторном - развитии. Они позже начинают ходить и сидеть. Нередко у них отменяются врожденные пороки сердца, характерные изменения дерматоглифики (на ладони имеется глубокая "обезьянья борозда").
Умственное развитие больных с синдромом Дауна отстает; если не применяются специальные методы обучения возможно, развитие тяжелой идиотии. Болезнь сопровождается расстройством эндокринных желез, нарушением обмена веществ, снижением иммунитета. По этой причине они часто болеют пневмонией, инфекционными заболеваниями (см. приложение табл. 4).
Продолжительность жизни больных ограничена. Однако с помощью специальных методов обучения, укрепления физического здоровья, правильного питания и ухода, применения лекарственных препаратов и др. ее можно значительно продлить. Многие люди с синдромом Дауна способны жить самостоятельно
Вместе с этим смотрят:
РЖсторiя виникнення та розвитку масажу
Аборты
Аденовирусная инфекция
Азотные и кислородные ванны, нафталановая нефть
Акушерська операцiя - накладання акушерських щипцiв